Optional exercises #1 :
Comparing divergent proteins with similar structures
load 2DLN.pdb load 1GSA.pdb
Are these structures similar? homologous?
Does Magic fit work?
For both structures Display the CA backbone only and color according to secondary structure. Use the layer info panel and orient the two structures so that they look similar.
Select the ADP molecules only
Use the 3 point alignment approach to align the ADP molecules
make the whole molecules visible again
move one structure over to the right (no turning)
select the ADP molecules
In the control panel header click on the cloud icon to display both ADPs in space filling mode.
In display click on render Q3D.
(To get more spectacular displays, you can save the pictures as POV files and use the program POV ray to make even nicer images)
If you have time, do the same for 1GSA, 2DLN and CPSBfrag and CPSFfrag . The latter two files are clippings of the front and back ATP binding sites of the carbamoyl phosphate synthetase (1BXR).
WHAT DOES THIS MEAN? Recall the use of 2DLN in PSI blast. Are all of these structures homologs? What does that tell you about evolution of proteins? An illustration is here .
Optional exercises #2 :
If you have more time to spare and you are up for a challenge, take a look at the nucleosome. Right click here and save as pdb file. Open it from within spdbv. You might want to do some of the future exercises with the nucleosome in addition to the ATPases - thus save the pdb file, where you can find it again.
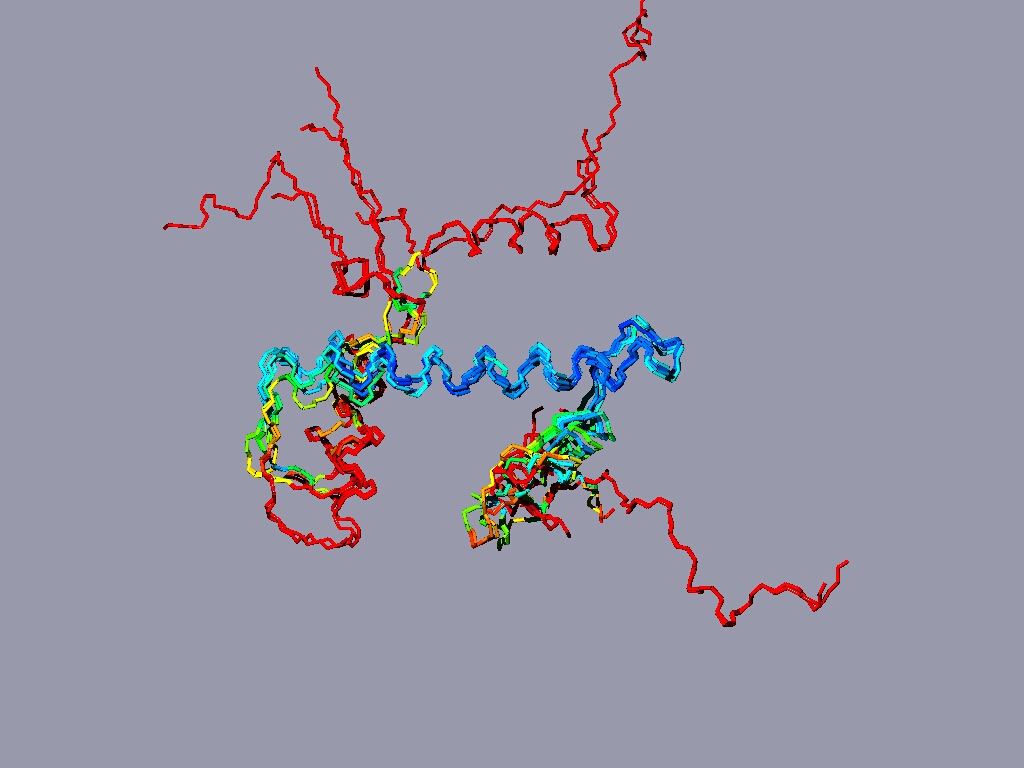
Align all the histones form the nucleosome to one reference histone and color in rmv:
The result might look something like this:
The picture shows a structure alignment of the 8 histones (2 each) that are part of the nucleosome. All the histones were colored regarding the match to H2A, except H2A, which was colored according to its match to H3. Coloring option RMS - shorter wavelengths - better match
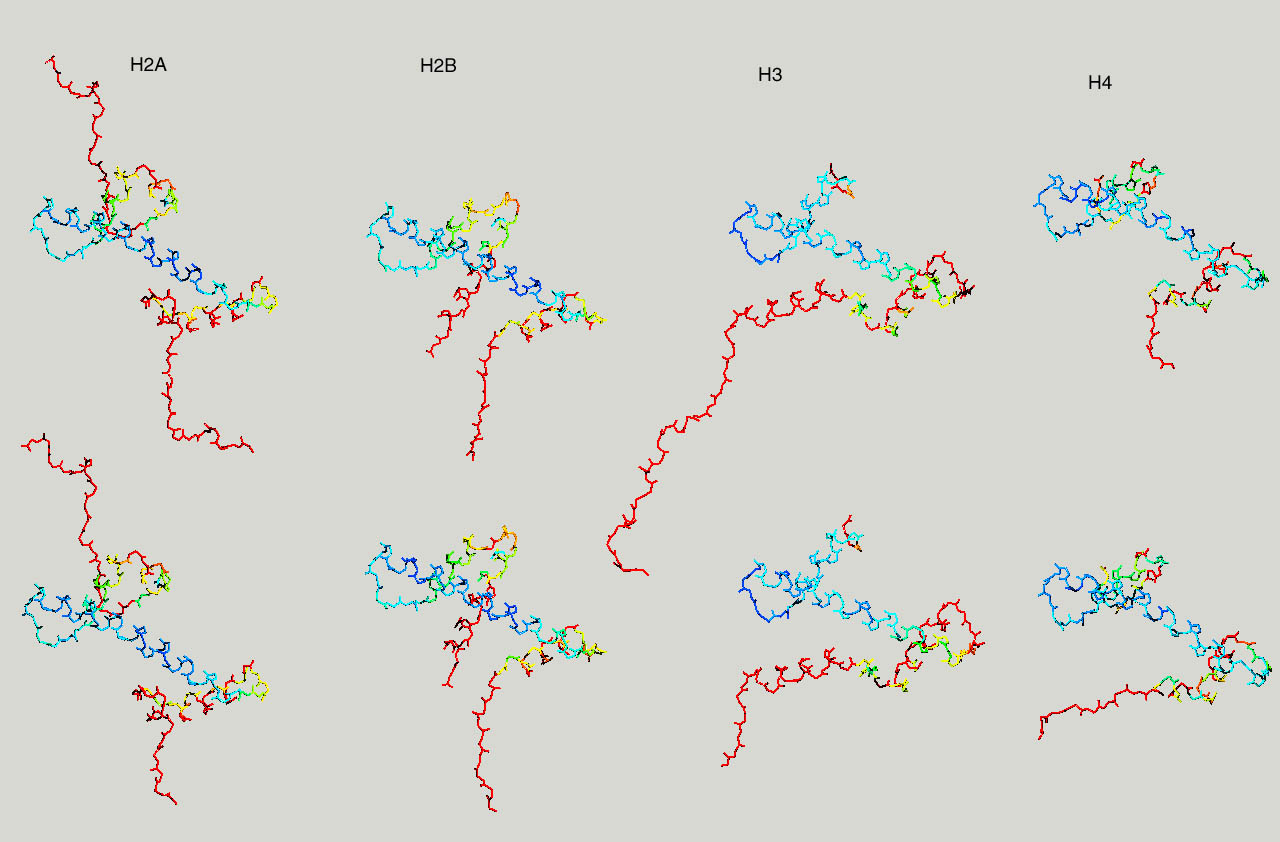
Below same as last figure, but histones are depicted side by side :
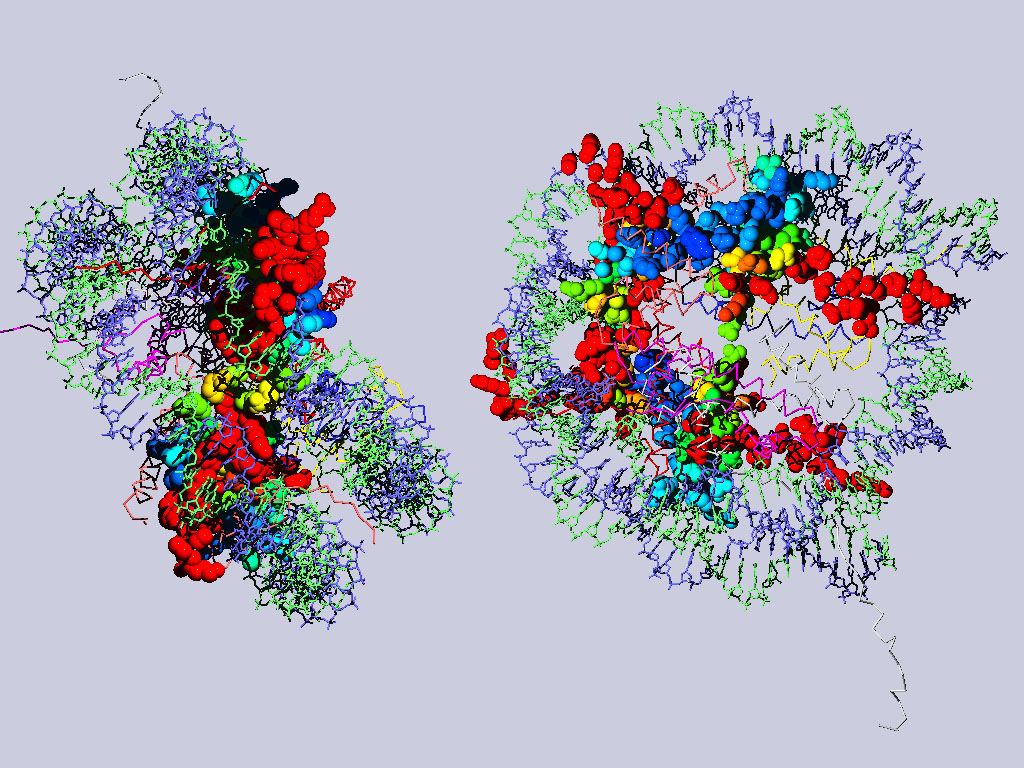
Below are two views of the complete nucleosome. Histones H2A are depicted as space filling balls and RMS colored regarding their match to H3. The rest of the molecule is colored according to chain.