CLASS 15. Introns and methods of gene prediction in Eukaryotes.
Two articles: [1] and [2] (Moodle);
Sections 9.3-9.6 (supp.textbook);
Sections 10.4-10.7 (supp. textbook) [OPTIONAL]
Introns and Their Evolution
Three groups of introns based on their splicing mechanisms:
group I and II are self-splicing: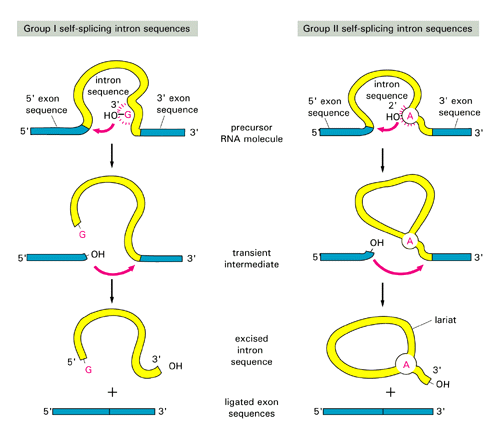
group III introns are present in eukaryotic nucleus, need spliceosomes to splice out:
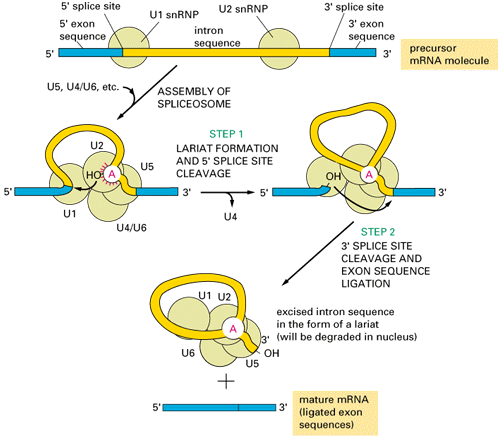
Where different groups of introns occur?
- Group I: were discovered in ciliated protozoan Tetrahymena; found also in Physarum, fungal mitochondria and phage T4
- Group II: common in Bacteria, and so far found only in one Archaeal genus, Methanosarcina
- Group III: present throughout eukaryotes, but more common in "crown-group" eukaryotes
Where do spliceosomal introns come from and how the splicing machinery evolved?
Hypothesis:
Spliceosomal introns evolved from Class II introns; the function of some of the internal loops of the class II introns are taken over by the spliceosomal snRNA (small nuclear RNA).Support:
- Group II introns are often located in intergenic regions in Bacteria, suggesting their mobility as parasitic genetic elements
- Group II and spliceosomal introns both form a lariat structure (see figures above)
- class II introns that are non-functioning because a loop has been removed splice in the presence of snRNA.
- The reverse is true too: domain of a group II intron can substitute snRNA of the spliceosome
Problem:
- class II introns are found in bacteria, and only in one Archaeal genus, Methanosarcina; why is it that predominately "crown-group" eukaryotes have introns?
How did the spliceosomal machinery evolve?
Gratuitous complexity hypothesis for evolution of spliceosomal machinery: See reading assignment on Moodle [the portions for the reading are highlighted in the PDF file].
This hypothesis stems from the principle that once a simple mechanism becomes more complicated through random change it is impossible to return to the less complex mechanism even if the introduced complexity contributes nothing and takes up more energy than the simpler process.
Consider this situation as an example:
Domain 5 of Group II introns is highly conserved and crucial to the self-splicing process. It is possible that at some point in evolution this domain acquired a defect and self-splicing could no longer occur. Somehow a protein complex (U6/U2 helix) rescued this function and a functional protein could once again be transcribed! This is more complicated and requires more energy but it cannot be reversed and it is therefore permanently more complex.
Intron Mobility
Introns in group II are able to insert into both homologous sites and ectopic sites via retrohoming and retrotransposition respectively. To learn more about the process by which group II introns insert into intronless sites, go here. Both group I and group II introns can have homing endonucleases. See the excellent paper by Goddard and Burt on the reinvasion cycle. Also: reverse splicing
Possible benefits of having introns:
They waste so much energy! Why aren't these junk segments of DNA purged from the genome?
Exon shuffling, alternative splicing (one gene -> several different protein products) ....
Two rival hypotheses: Intron Early vs. Intron Late
Intron early:
Protein diversity arose in analogy to exon shuffling in the generation of antibody diversity (see your biochemistry or genetics textbook on the maturation of the immune system).Claims:
- Introns separate structural domains. Example of a Go-plot is here (horse haemoglobin; from here, these authors describe a significant excess of introns in the linker regions defined through the overlap in the Go-plot).
- Introns arose early, before the uptake of the mitochondrial and chloroplast endosymbiont.
- Neighboring introns often are in the same phase. While significant, the excess is rather small. However, the excess is larger, if only multidomain proteins are considered, suggesting that these indeed evolved through exon shuffling (see here for a recent analysis).
Intron late:
Present day introns are late invaders of already functional genes. Exon shuffling might play some role in eukaryotes, but most of protein diversity arose before introns invaded protein coding genes.Claims:
- distribution of introns mapped on phylogentic trees unambiguously points towards late invasion (and here).
- The correlation between structure and intron position is not unambiguous.
- The finding that mitochondrial (bacterial) and nucleocytoplasmic genes have introns in the same location could reflect a preferred intron integration site. The phase pattern is also observed in vertebrate genes, in which the introns are of late origin.
- Exon shuffling requires introns located in the same phase, but there might be other reasons for having a slight excess of introns in the same phase. For introns to frequently invade genes, there needs to be mechanisms for introns to find new "homes" (see above).
Compromise:
mixed model of intron evolution- version 1 - while some introns are recent, most are old. E.g.: [Roy, 2003].
- version 2 - while most introns are recent, some are older, but not necessarily very old. E.g.: [Rogozin et al., 2003]
Prediction of genes in Eukaryotes
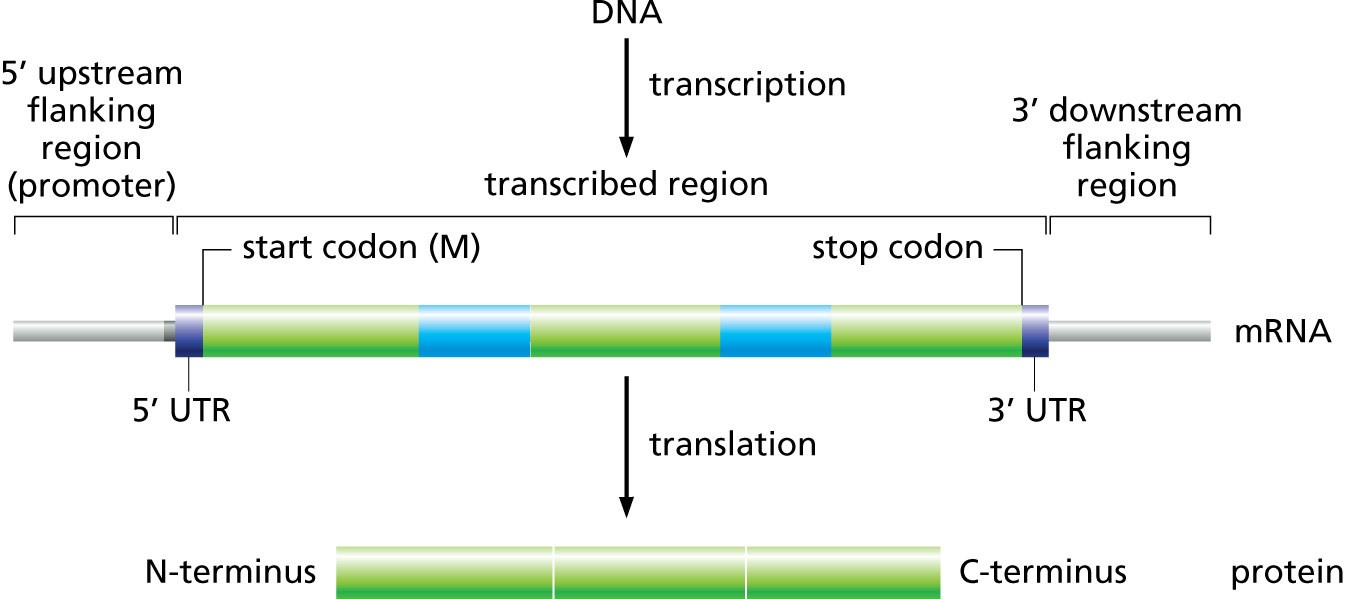
Challenges to gene prediction in Eukaryotes:
- Lower gene density in general (with regions of lower and higher gene densities, corresponding to isochores).
- Presence of introns means that protein-coding portions (exons) are on average shorter than in prokaryotes.
- Correct reading frame needs to preserved when joining exons.
- Alternative Splicing:
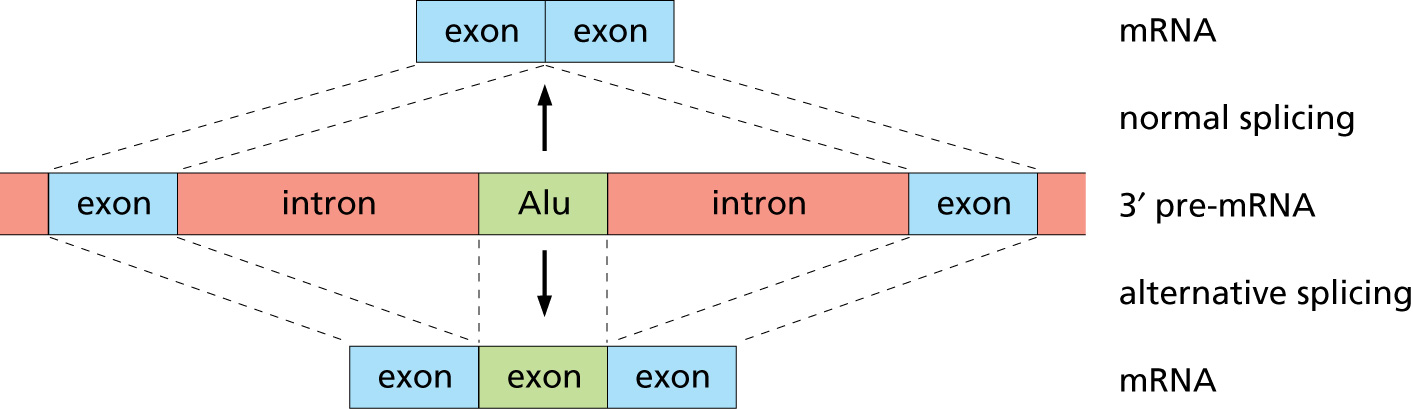 Fig. B9.5 [Source]. Example of Alternative Splicing.
Fig. B9.5 [Source]. Example of Alternative Splicing.
-
Not much of a splice site consensus ("exon1 GT"-"intron"-"AT exon2").
 Fig. Splice site consensus in Arabidopsis thaliana (model plant species). This table summarizes the sequences surrounding the intron splice sites in the plant Arabidopsis. E.g., in 52.9% of the intron exon boundaries (bottom part) the first base of the exon is a G, and in 40.5% the next nucletides is a T.
Fig. Splice site consensus in Arabidopsis thaliana (model plant species). This table summarizes the sequences surrounding the intron splice sites in the plant Arabidopsis. E.g., in 52.9% of the intron exon boundaries (bottom part) the first base of the exon is a G, and in 40.5% the next nucletides is a T.
[Source]
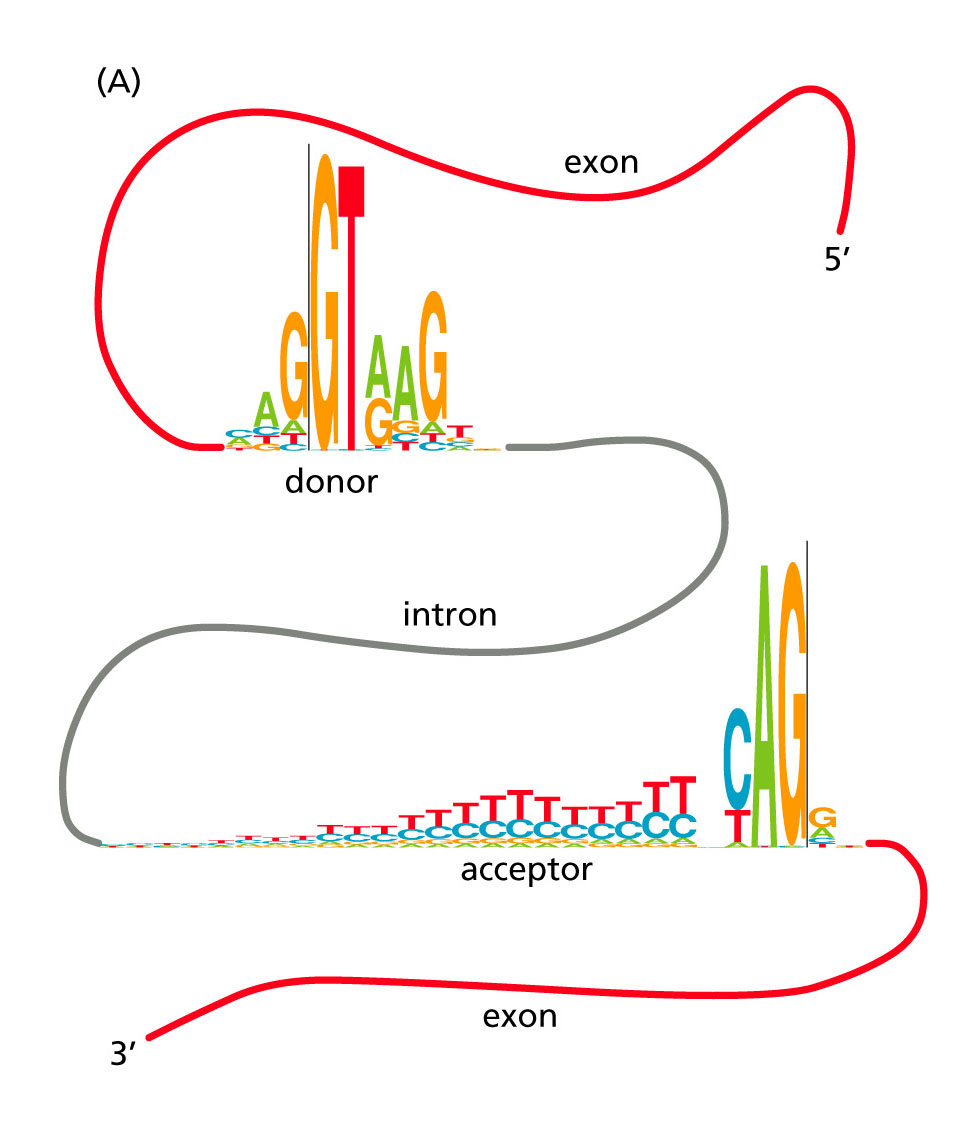 Fig. 10.11A [Source]. Conservation of intron splice-site signals in human.
Fig. 10.11A [Source]. Conservation of intron splice-site signals in human.
- Eukaryotic promoters are more complex than their prokaryotic counterparts: e.g., they may be located thousands nucleotides away from transcription start site, can be either upstream or downstream of the gene and more complex in structure.
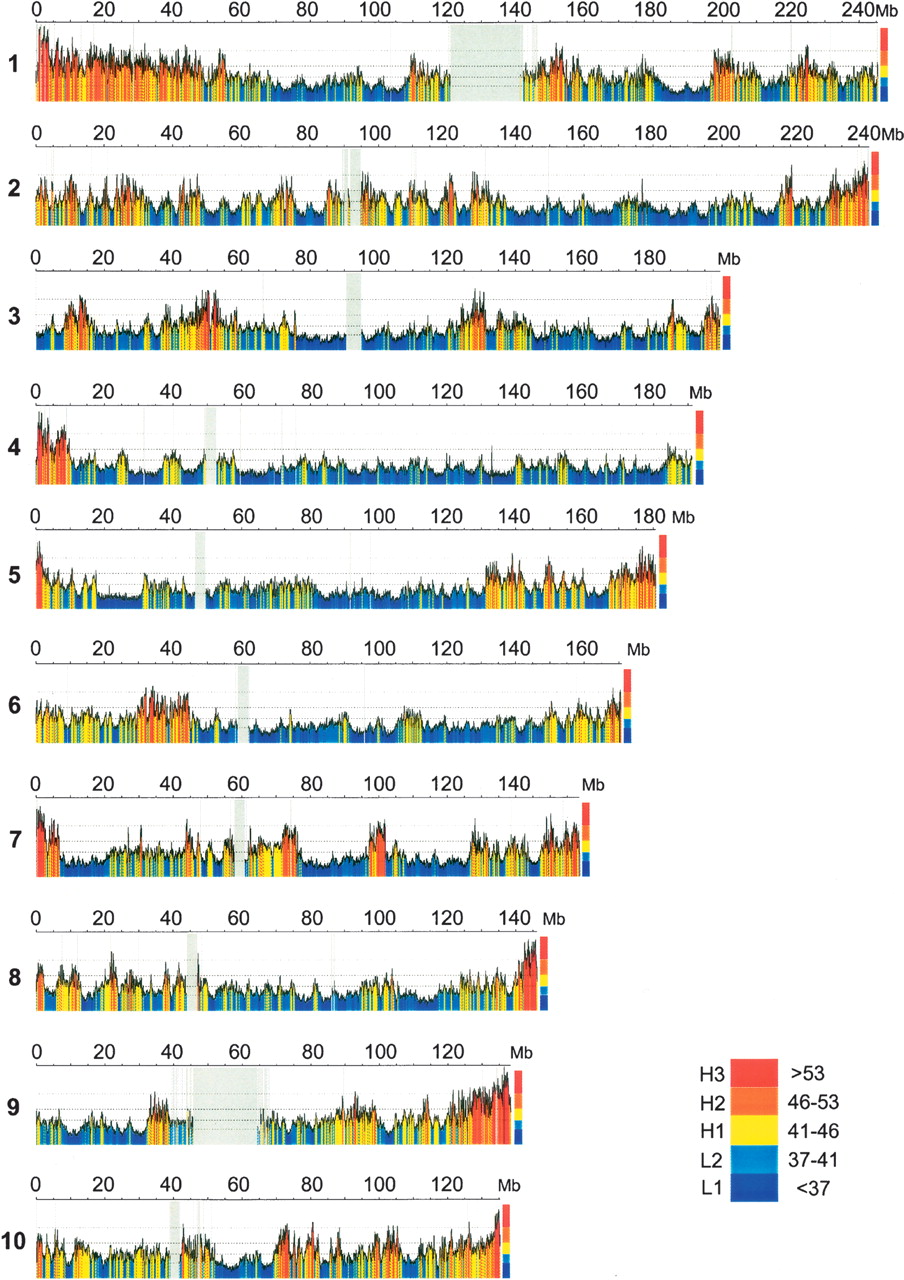
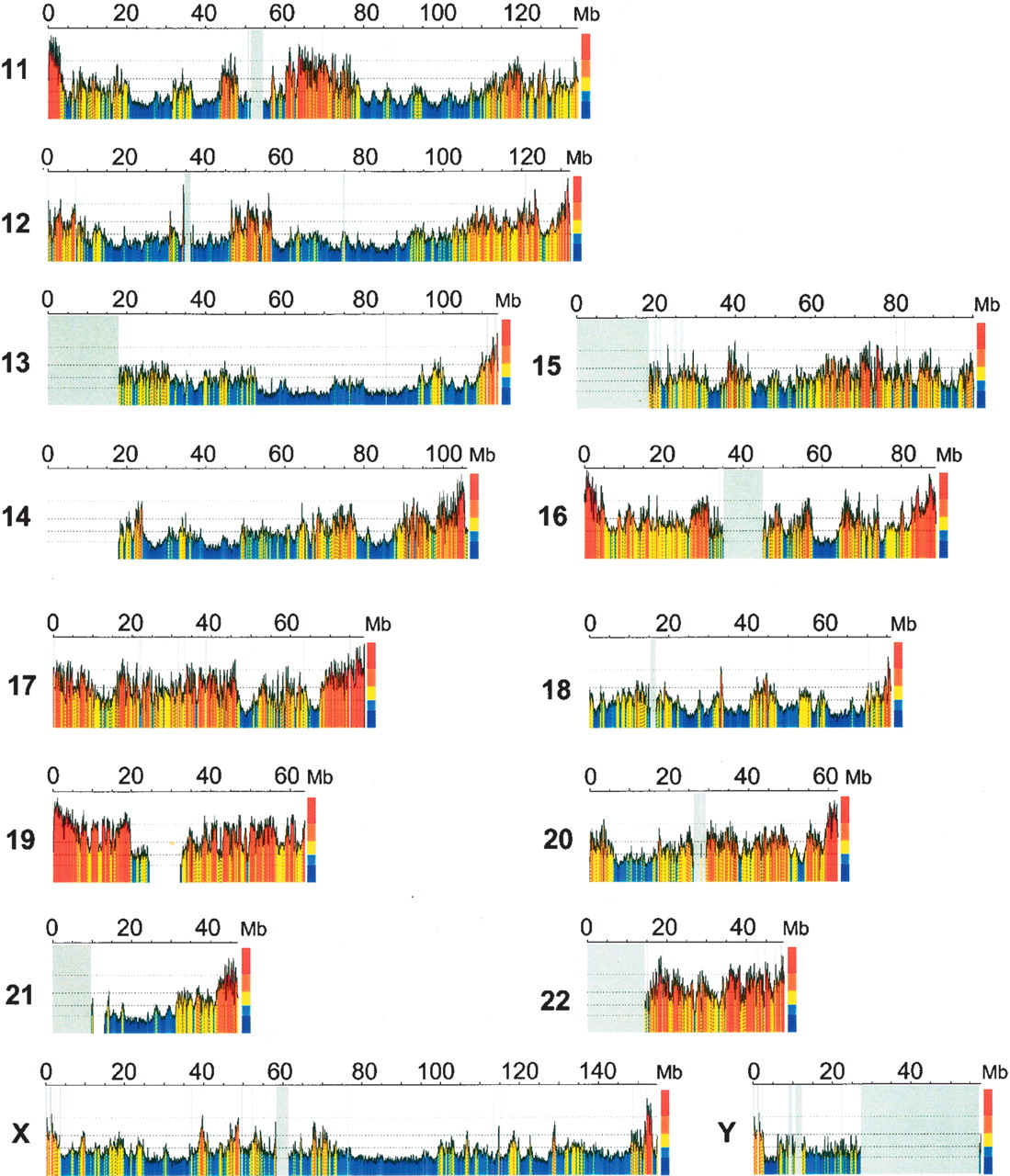
One of the most accurate programs (see Table 8.2 in your textbook) is GenScan. It uses 5-th order HMMs (hexamers).
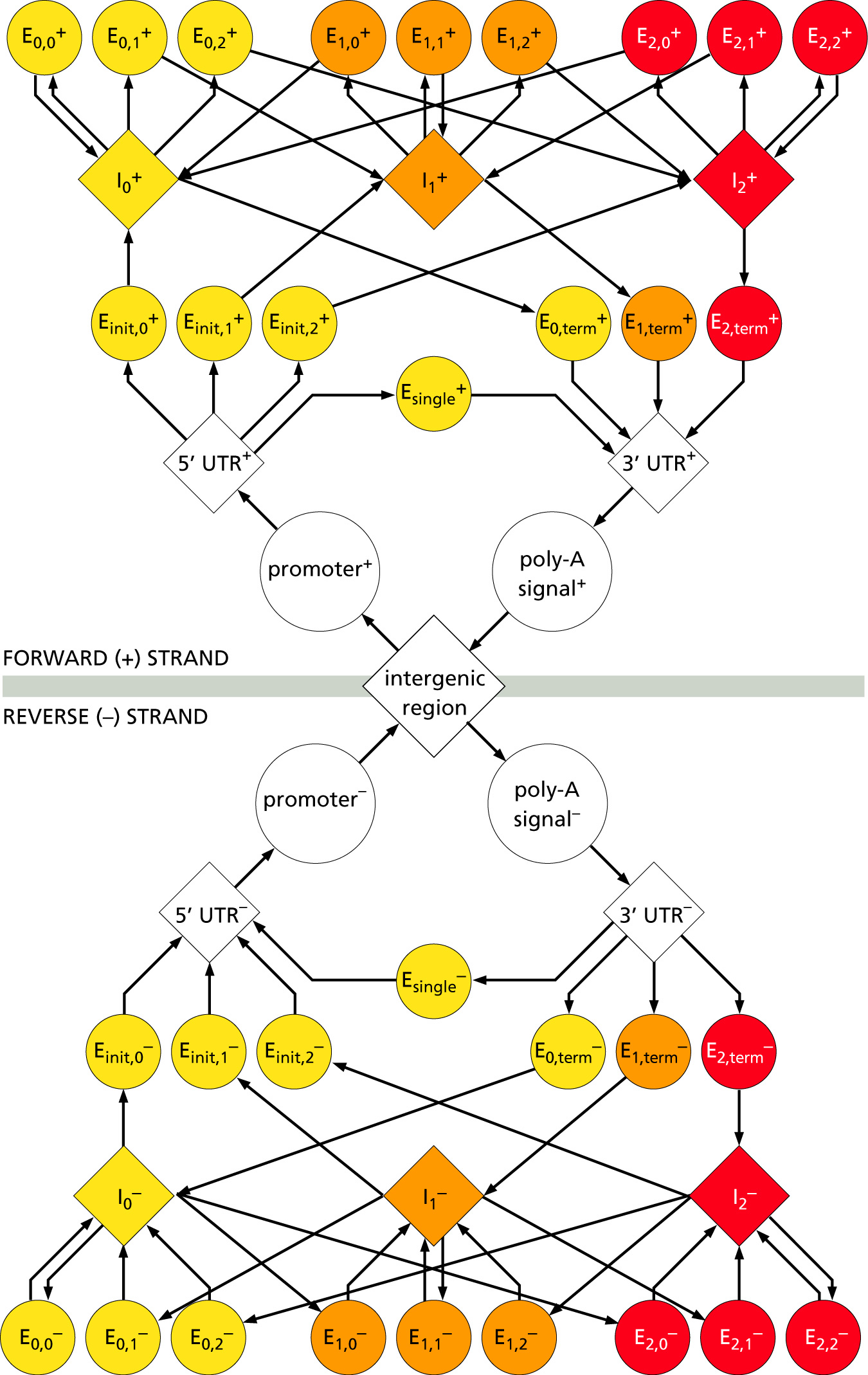
Putative exons are assigned a probability score (P). The program was designed primarily to predict genes in human/vertebrate genomic sequences.
GenomeScan combines GenScan with BLASTX searches.