CLASS 29. Distance, Parsimony and ML phylogenetic analyses. Long Branch Attraction.
IMPORTANT ANNOUNCEMENT: CLASSES FOR MARCH 22,24 and 26 are CANCELLED.
HOME WORK ASSIGNMENT: READ CH. 17 (textbook), FINISH THIS LAB (BELOW), WORK ON ANNOTATHON PROJECT AND GO THROUGH PROBLEM SET #4.
HOMEWORK PROBLEM SET #4 is posted to Moodle.
For each exercise, provide search query used and keep the answers brief. Email me the answers by Sunday, March 28 11:59PM AST at the latest.
Use "CLASS 29 EXERCISE" as a message subject, and type answers directly to email body (i.e., no document attachments please). Make sure that first line of your message is your NAME.
PART 1. Bootstrapping.
Bootstrap analyses using neighbor joining method (ClustalX).
Load these sequences into ClustalX, align them using the default options.
In the Trees menu select "correct for multiple substitutions" and "exclude positions with gaps" (there should be checkmarks visible in the menu, before you run start the bootstrap analysis).
Calculate "Bootstrap NJ tree". The results will be written into a tree with ".phb" extension.
Inspect the tree using NJPlot. Root the tree as appropriate and check display bootstrap support values.
Which branches have only low bootstrap support? Are any of these findings surprising? What might be the reason?- Bootstrap analyses using parsimony (PHYLIP).
While still in your ClustalX alignment:
File -- Save Sequences as...
tick PHYLIP format
This will generate a file ending in ".phy" with your alignment in PHYLIP format.
Edit this ATPaseSU.phy file, changing all gap "-" characters to missing data "?" characters (use MS Word or any other text editor).
Download PHYLIP
Copy this ATPaseSU.phy file into the phylip-3.69/exe folder
Go into the "phylip-3.69" folder, then into "exe" subfolder
Run "seqboot"seqboot: can't find input file "infile" Please enter a new file name> ATPaseSU.phy Y Random number seed (must be odd)? an-odd-number completed replicate number 10 completed replicate number 20 completed replicate number 30 completed replicate number 40 completed replicate number 50 completed replicate number 60 completed replicate number 70 completed replicate number 80 completed replicate number 90 completed replicate number 100 Output written to file "outfile" Done.
Rename "outfile" to ATPaseSUboot.phy
Run "protpars"
protpars: can't find input file "infile" Please enter a new file name> ATPaseSUboot.phy j (Randomize input order of sequences) Random number seed (must be odd)? an-odd-number Number of times to jumble? 1 m (Analyze multiple data sets) Multiple data sets or multiple weights? (type D or W) d How many data sets? 100 Random number seed (must be odd)? 1 Number of times to jumble? 1 y (go!)
Rename outtree to ATPaseSUboot.tre
Run "consense"
consense: can't find input tree file "intree" Please enter a new file name> ATPaseSUboot.tre consense: the file "outfile" that you wanted to use as output file already exists. Do you want to Replace it, Append to it, write to a new File, or Quit? (please type R, A, F, or Q) R Are these settings correct? (type Y or the letter for one to change) y Consensus tree written to file "outtree" Output written to file "outfile" Done.
Rename outtree to ATPaseSUconsensus.tre
Inspect the tree using njplot. Root the trees as appropriate and check "display bootstrap support values". Which branches have only low bootstrap support? Very briefly, how does this compare to the previous tree made with ClustalX using a distance-based analysis? - Bootstrap analyses using maximum likelihood (PhyML). This takes too long to be finished in class, so this analysis is optional (to do at home). Use PhyML Online to reconstruct ML tree and compare it to the trees obtained in previous two exercises.
PART 2. LONG BRANCH ATTRACTION.
- Long Branch Attraction (LBA) is a serious problem in phylogenetic reconstruction. LBA denotes the fact that long branches tend to be grouped together with significant support, even though the organisms representing the long branches did not share more recent common ancestry. The support usually is measured through bootstrap support values for the different trees. The evolution of four sequences (named A,B,C,D) was simulated according to the following tree:
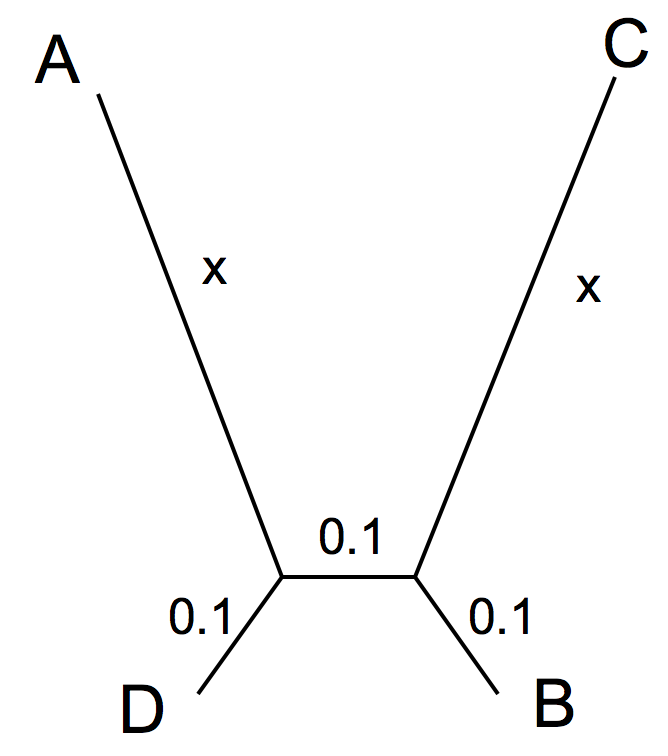
Files containing these sequences in multiple sequence fasta format were generated and named according to the length (x) chosen for the two long branches (all scaled in substitutions per site). For the simulation we assumed that the among-site-rate-variation could be described with a gamma distribution that has a shape parameter of 1 (equivalent to an exponential distribution).
These files are HERE. Save the files sequences in the directory where the PHYLIP executables are stored (within the "phylip-3.69/exe" folder).
Your task is to explore the sensitivity of different phylogenetic reconstruction algorithms towards LBA. In this case we know that the sequences are aligned as given (since insertions/deletions were not simulated), i.e., you could just load them into clustalx and save them as phylip-formatted files. Alternatively, you could explore the effect that the alignment algorithm has on LBA. To keep track of things, name the files accordingly.
To test parsimony, choose the files with x = 0.1, 0.3, 1, 3. Generate the corresponding files in Phylip format using ClustalX.
How long are the sequences (if you aligned them, how long are they after the alignment)?
Use SEQBOOT from the PHYLIP package to generate 100 bootstrap samples for each file. By default PHYLIP writes these into a single multiple sequence file. This file is named outfile. Rename it into something that reminds you of what is in this file (e.g.: 0_1.phy.boot).
Use PROTPARS from the PHYLIP package to evaluate the 100 bootstrap samples (select option M for multiple files). The program generates a file that contains the trees calculated from each sample in Newick format (outtree). Rename this file (e.g., 0_1.boot.protparstrees).
Use the CONSENSE program form the PHYLIP package to calculate the consensus tree and its bootstrap support values.
As an answer, list the files that you choose, aligned or as provided, the bootstrap support for the correct tree, and the support for the LBA tree.
Explore performance of distance methods with respect to LBA. Depending on the settings, these might be less sensitive to LBA. x = 0.3, 3, 30, 300 are good choices to explore.
To do the analyses in PHYLIP:
Convert the sequences to PHYLIP format using clustalx. (convert only and/or align the sequences (as above))- Load the sequences into SEQBOOT and generate 10 bootstrap samples.
- Load the bootstrap samples into PROTDIST (you need to choose option M), the JTT substitution matrix (default) is a good choice; also option G is recommended (if asked for the parameter choose 1, don't choose the option where you need to assign weights). The outfile of PROTDIST contains the distance matrices (rename the outfile).
- Load the distance matrices into FITCH or NEIGHBOR. Again select option M for multiple files (else the default values are reasonable.). Rename outtree
- Feed it into the CONSENSE program.
As an answer, list the parameters you chose for protdist, the files that you chose (aligned or as provided), and for each file indicate the bootstrap support for the correct tree, and the support for the LBA tree.
Explore how the sensitivity of Protdist towards LBA depends on the correction for multiple substitutions.
- Load the sequences into SEQBOOT and generate 10 bootstrap samples.
- Load the bootstrap samples into PROTDIST (again, you need to choose option M). Things to vary include the G-option with less ASRV (e.g., alpha =20); or you could choose a simple Kimura correction (this is what ClustalX uses to correct for multiple substitutions) by toggling the P option. The outfile of PROTDIST contains the distance matrices (rename the outfile).
- Load the distance matrices into FITCH or NEIGHBOR. Again select option M for multiple files (else the default values are reasonable). Rename outtree and
- Feed it into the CONSENSE program.
As your answer, give the parameters you chose for protdist, the files that you chose, indicate if you aligned them or used them as provided, and for each file give the bootstrap support for the correct tree, and the support for the LBA tree.