COMPARATIVE ANALYSIS OF THE DEINOCOCCUS GENOME
INDICATES
INFREQUENT HORIZONTAL TRANSFER BETWEEN THE THREE DOMAINS OF LIFE
Members of the deep branching bacterial lineage Deinococcaceae
(e.g. Thermus and Deinococcus) are classified as Bacteria based on
cell wall, lipid composition, and ribosomal RNA sequence (Hensel et al., 1986).
However, Thermus thermophilus does not have an F-ATPase
like other bacteria, but a complete A/V-ATPase (Yokoyama et al., 1990;
Tsutsumi et al., 1991). A-ATPases are usually found only in archaea while
V-ATPases are usually found only in eukaryotes. The typical ATPase of bacteria
is the F-ATPase, although A/V type ATPases have been found in spirochetes, Enterococcus,
and Chlamydia. The majority of Thermus and Deinococcus
species examined thus far contain only an A/V-ATPase, although a
recent report indicates that Thermus scotoductus contains an
F-ATPase (Radax et al., 1998). The vacuolar/archaeal type ATPase
genes are not the only evidence of non-bacterial type genes in the
Deinococcaceae. Thermus flavus was shown to have a malate
dehydrogenase that in phylogenetic analyses groups with the eukaryotic
homologues (Iwabe et al., 1989). Genetic screening of a Thermus ruber
genomic library with randomly primed probes made from
Thermoplasma acidophilus genomic DNA identified a
number of clones which hybridized to the archaeal probes.
One of these encoded a prolyl-tRNA synthetase, which was shown
to be more closely related to archael and eukaryotic type prolyl
tRNA synthetases than to the majority of bacterial prolyl tRNA
synthetases (Gogarten et al., 1999). The most plausible
explanation for the presence of non-bacterial genes in members of the
Deinococcaceae is horizontal gene transfer from the archaeal
lineage to this bacterial lineage (Olendzenski et al., 1998).
This hypothesis led us to use a bioinformatics
approach to search the Deinococcus radiodurans genome
for other candidate genes that might have been horizontally
transferred from the archaeal domain (Fig. 1).
The genome of D. radiodurans has been completely sequenced,
partially assembled, and is available in preliminary form through
the TIGR web-page. Putative open
reading frames (ORFs) from the Deinococcus genome were searched
against representative completed genomes from each of the three
domains of life. E values for each ORF obtained using BLAST
against the yeast, E. coli and Methanococcus genomes
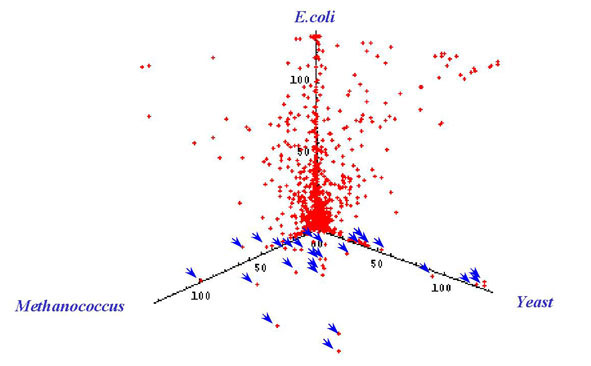
where plotted on a three dimensional graph (Fig. 2),
allowing visualization of genes that are likely candidates for
horizontally transferred genes. Those ORFs that showed highest
matches against archaeal and eukaryotic genes were collected and
ranked (Table 1). Among the top ranked hits were the A/V-ATPase
catalytic and non-catalytic subunits, (Fig. 3) and the prolyl-tRNA
synthetase (Fig. 4). Additionally, the high scores obtained in
searches against the Saccharomyces, Methanococcus,
and Escherichia genomes were plotted in a 3-D graph (Fig. 5).
Putatively frequently transferred genes are those which reside on or
close to the line which is equally distant from all three axes and
are depicted as red dots (Fig. 5, and Table 2).
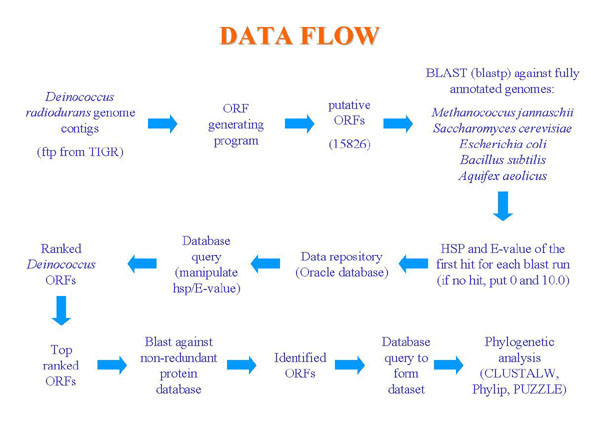
Figure 1. Data Flow. Steps of analysis for determining potential horizontally transferred genes.
Figure 2. Plot of E-values for Deinococcus genes obtained using BLAST searches against the genomes of Yeast, E. coli, Methanococcus. Each point (x, y, z) represents one putative ORF from the Deinococcus genome. The axes are as follows: x is -log E-value from BLAST against the yeast genome; y is -log E-value from BLAST against the Methanococcus genome; z is -log E-value from BLAST against the E. coli genome. The arrows indicate the points which are much more similar to Yeast and/or Methanococcus genes but much less similar to E. coli genes based on the E-value and represent candidate genes horizontally transferred from Archaea, or Eucarya to the Bacterial domain.
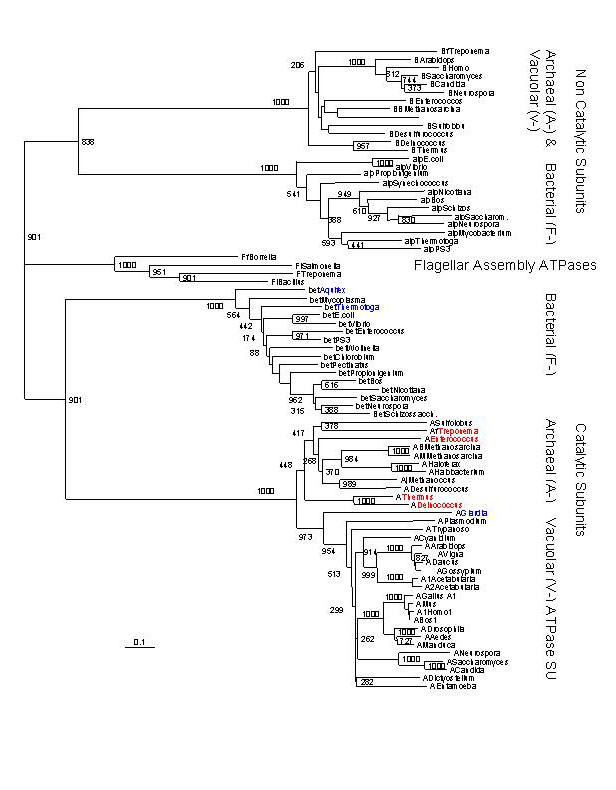 Fig. 3. Distance tree based on amino acid alignment of catalytic and non-catalytic subunits of proton pumping ATPases from Bacteria, Archaea and Eukaryotes. The catalytic and non-catalytic subunits are the products of an ancient gene duplication; the tree can be rooted by using one of the duplicated pair (paralog) as an outgroup. The genera in red are Bacterial species (including Thermus and Deinococcus) which have aquired an Archeal type ATPase. The genera in blue represent the deepest branches in the Bacterial and Eukaryotic lineages.
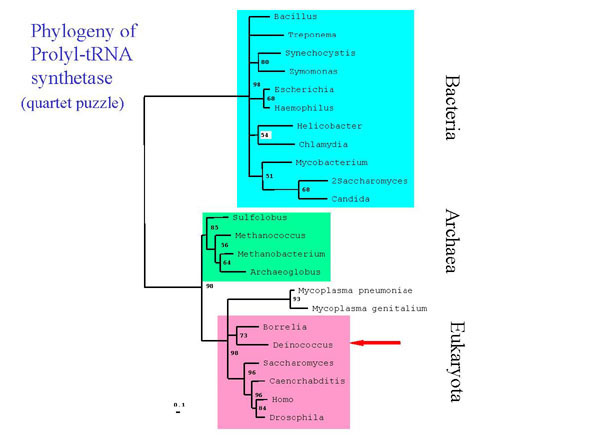 Fig. 4. Quartet puzzling analysis of prolyl tRNA synthtase amino acid sequences. Sequences were aligned using the profile alignment option of ClustalX with default parameters. Numbers reflect quartet puzzling support values for each node. Nodes with values of less than 50% have been collapsed. Analysis was corrected for among site rate variation using the Gamma distribution(=1.97). All sequences were obtained from GenBank except those of Deinococcus and Treponema, which were obtained through BLAST search from TIGR, and Sulfolobus, which was kindly provided by Mark Regan and Cristoph Sensen, Institute for Marine Biosciences, Dalhousie University, Halifax, Nova Scotia.
Deinococcus and Thermus (not shown) contain a prolyl TRNA synthetase that is more closely related to those found in Archea and Eukaryotes than in the majority of other Bacteria. This result was obtained both through library screening of a Thermus library with Archeal probes and by using the described bioinformatic approach. The Deinococcacea are not the only bacteria that seem to have aquired an Archeal type prolyl tRNA synthetase by horizontal gene transfer- The spirochete Borrelia and the genus Mycoplasma also have this type. The yeast homologs which group among the Bacteria (2Saccharomyces and Candida) are assumed to be of mitochondrial origin.
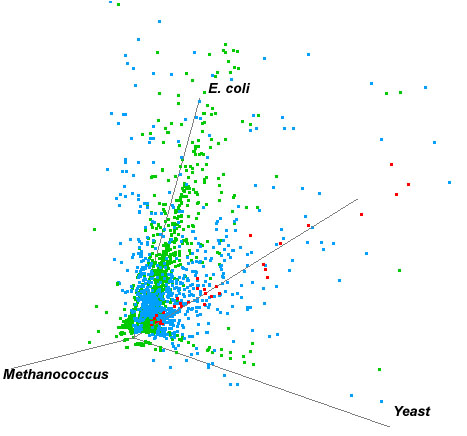 Figure 5. 3-D Plot of High Scores obtained in BLAST search against the Saccharomyces, Methanococcus and Escherichia genomes. Red dots represent the genes which reside on or close to the line which is equally distant from all three axes. These represent genes which potentially have been frequently transferred throughout evolutionary history and thus have HSP values which are more or less equivalent among each of the three domains.
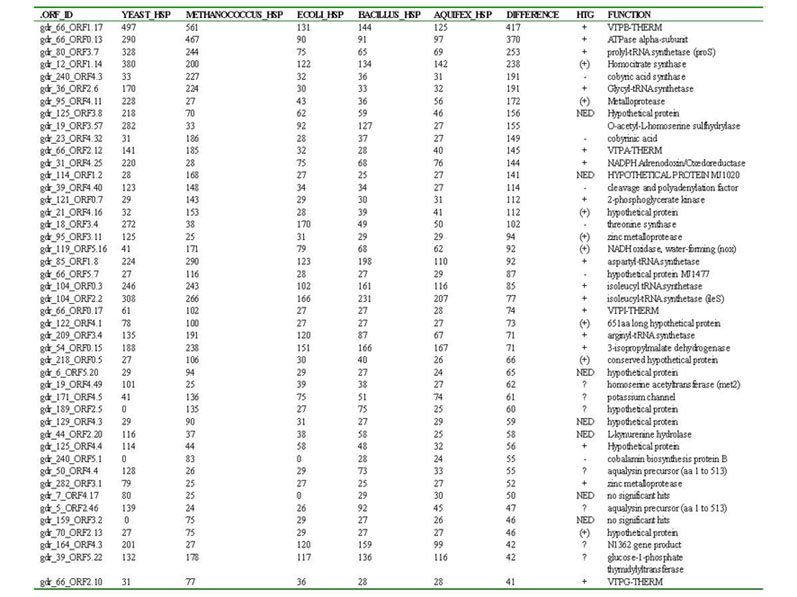 Table 1. The top 45 HSP hits between Deinococcus and Yeast, Methanococcus, E. coli, Bacillus, Aquifex. For each ORF, datasets of homologous amino acid sequences were constructed and analyzed using distance and/or quartet puzzling analyses. Based on the resulting phylogenetic analyses, each was classified as to whether or not it was transferred horizontally from Archaea or Eukaryotes to Deinococcus. These results are summarized in the HTG column. Symbols are as follows: + = tree toplogy differs from that of the three domains, and suggests horizontal transfer, (+) = tree toplogy suggests potential horizontal transfer, - = tree topology does not suggest horizontal transfer (i.e. tree reflects three domains), NED = not enough data to analyze, ? = unable to analyze reliably due to numerous paralogs in data set, or highly divergent Deinococcus sequences which could produce long branch artifacts.
 Table 2.
Identity of sequences depicted as red dots in Fig.5, including ORF ID and HSP(High Score Pair) values between Deinococcus and Yeast, Methanococcus, E. coli, Bacillus, or Aquifex. Functions listed are based on the highest hits in BLAST analysis. These sequences represent genes that have potentially been transferred frequently among the three domains.
- The computer method based on homologues search among several completed microbial genomes provides good initial screen for the candidates of horizontally transferred genes. This method may apply to the analysis of other microbial genomes.
- v A number of candidate genes are found, including confirmation of genes that were already known to be horizontally transferred, namely the ATPase subunits and prolyl-tRNA synthetase which had been discovered by a genomic library hybridization method.
- In comparison to the total number of genes in Deinococcus, the number of candidate genes horizontally transferred from Archaea to Deinococcus genomes is very limited, suggesting that this type of gene transfer did not happen very frequently in the past or to a large extent in the history of this lineage.
We gratefully acknowledge the contributions of the following people:
Ryan D. Murphey,
Dept. of Molecular & Cell Biology
for his work in screening the Thermus library with archeal probes.
Dr. Dong-Guk Shin,
Dept. of Computer Science & Engineering
for computing facilities including use of the Oracle database.
- Hensel, R., Demharter, W., Kandler, O., Kroppenstedt, M., Stackebrandt, E.:1986, Internatl. J. Syst. Bact. 36, 444-453
- Iwabe, N., Kuna, K.I., Hasegawa, M., Osawa, S., Miyata, T.: 1989, Proc. Natl. Acad. Sci., USA 86, 9355-9359
- Olendzenski, L., Hilario, E., Gogarten, J.P.: 1998, In: Horizontal Gene Transfer, Chapman and Hall, London. pp 349-362
- Radax, C., Sigurdsson, O., Hreggvidsson, G.O., Aichinger, N., Gruber, C., Kristjansson, J.K., Stan-Lotter, H.: 1998, Syst. Appl. Microbiol. 21, 12-22
- Tsutsumi, S., Denda, K., Yokoyama, K., Oshima, T., Date, T., Yoshida, M.: 1991, Biochim. Biophys. Acta, 1098, 13-20
- Yokoyama, K., Oshima, T., Yoshida, M.: 1990, J. Biol. Chem., 265, 21946-21950
- Gogarten, J.P., Murphey, R.D., Olendzenski, L.:1999, Biol. Bull. 196:359-362
|
|