GOALS:
·
Learn how to get your data into TREE-PUZZLE and the results out.
·
Attain some understanding on
what to do with the ml ratio test.
·
Think about models for the substitution
process.
·
Learn about the different options
in TREE-PUZZLE
General Remarks:
If no file called
infile is present in the program directory, TREE-PUZZLE will prompt you for the
infile. If you work a lot on the same
dataset, it might be worth to rename your data set into infile. The output is always put into the files outfile,
outtree and outdist.
You have to rename these files as soon as you are done. The next time TREE-PUZZLE starts, the files get erased (this is particularly
sad if your run took a couple of days).
The following assumes that you read the notes regarding TREE-PUZZLE from class 10. If you have not done so, do it now (here are the sections on puzzle trees, the ml-ratio test and ml-mapping).
![]() Open
this page in NETSCAPE and click HERE. When prompted
save the program TREE-PUZZLE.exe into your work folder.
Open
this page in NETSCAPE and click HERE. When prompted
save the program TREE-PUZZLE.exe into your work folder.
![]() Transfer the files testseq5.txt, testseq5.aln and testseq5.phb
onto your computer. These files contains
vacuolar/archaeal ATPases from 49 pro- and eukaryotes:
Transfer the files testseq5.txt, testseq5.aln and testseq5.phb
onto your computer. These files contains
vacuolar/archaeal ATPases from 49 pro- and eukaryotes:
Daucus
carota,
Arabidopsis thaliana, Gossypium hirsutum are plants;
Acetabularia acetabulum is a green
and Cyanidium caldarium is a red algae
Mus, Homo, Bos (mammals)
Gallus (bird), Drosophila, Aedes (insects), Ascidia (tunicate,
chordate) are animals
Saccharomyces, Candida, Schizosaccharomyces,
Eremothecium and Neurospora are
fungi
Dictyostelium discoideum, Entamoeba,
Plasmodium falsiparum, Trypanosoma, Nosema, and Giardia
are protists or protozoa
Sulfolobus acidocaldarius (70
°C), Sulfolobus solfataricus (70-85 °C), Archaeoglobus
fulgidus (83 °C), Methanosarcina barkeri (30-37 °C), Methanosarcina mazeii
(37°C),
Methanococcus jannaschii (80°C), Haloferax volcanii
(37°C),
Halobacterium salinarium (ca37°C), Methanobacterium
thermoautotrophicum (60-65°C), Desulfurococcus sp. (85-90 °C), Thermococcus sp
(75+ °C),
Aeropyrum pernix (90°C), Thermoplasma acidophilum (55-60°C), Pyrococcus
abysii and Pyrococcus horikoshii 95°C (archaea),
Enterococcus hirae (37°C), Borrelia
burgdorferi (33-37°C), Thermus thermophilus (70-80°C),
Deinococcus radiodurans (30°C), Chlamydia trachomatis,
Chlamydophila pneumoniae (Bacteria) are prokaryotes.
(Usually Bacteria have an F- and not an A-ATPase. The bacteria probably obtained
the archaeal/vacuolar type ATPase through horizontal gene transfer.)
Note
the German Collection of Microorganisms and
Cell Cultures is a good place to find growth temperatures for microorganisms.
The
prokaryotes can be considered as outgroup for the eukaryotes.
![]() Use maximum likelihood mapping to address the following
question: Does the Giardia or the Trichomonas sequence represent the deeper branching lineage among the eukaryotic homologues?
Use maximum likelihood mapping to address the following
question: Does the Giardia or the Trichomonas sequence represent the deeper branching lineage among the eukaryotic homologues?
As we are not sure about the relative branching order of Trypanosoma and Plasmodium,
do not assign them to any group.
For this you need to:
![]() load testseq5.aln into clustalx,
load testseq5.aln into clustalx,
![]() save the sequences (they should be
already aligned) in phylip format. Check
that truncating the sequence names does not result in duplicate sequence names.
save the sequences (they should be
already aligned) in phylip format. Check
that truncating the sequence names does not result in duplicate sequence names.
![]() Start TREE-PUZZLE by double clicking on the TREE-PUZZLE
icon
Start TREE-PUZZLE by double clicking on the TREE-PUZZLE
icon
![]() Load testseq5.phy (or what ever name you gave
the file)
Load testseq5.phy (or what ever name you gave
the file)
![]() Select type of analyses: maximum likelihood
mapping (“b”)
Select type of analyses: maximum likelihood
mapping (“b”)
![]() Select sequences in 4 clusters (“g”)
Select sequences in 4 clusters (“g”)
![]() Assign sequences to the 4 groups (i.e.
the 4 groups attached to the branch you want to study). Ask if you cannot figure it out.
Assign sequences to the 4 groups (i.e.
the 4 groups attached to the branch you want to study). Ask if you cannot figure it out.
![]() Select number of quartets (“n”) enter “0”
to select all (as two of your clusters contain a single sequence only, this
number is quite small)
Select number of quartets (“n”) enter “0”
to select all (as two of your clusters contain a single sequence only, this
number is quite small)
![]() Select the model of heterogeneity. (select Gamma distribution with 8 classes,
enter the shape parameter as 0.6. DO
NOT have the program find the shape parameter (takes about 30 minutes).
Select the model of heterogeneity. (select Gamma distribution with 8 classes,
enter the shape parameter as 0.6. DO
NOT have the program find the shape parameter (takes about 30 minutes).
![]() Interpret the outfiles.
Interpret the outfiles.
![]() What is your conclusion? (Which sequence
is deeper branch? Is the support for your finding strong, week, or nonexistent?)
What is your conclusion? (Which sequence
is deeper branch? Is the support for your finding strong, week, or nonexistent?)
![]() Select one of the following two problems to address
using the maximum likelihood ratio test:
Select one of the following two problems to address
using the maximum likelihood ratio test:
A)
Do the V-ATPase A subunits from the red
and green algae form a monophyletic group with the higher plants?
For
this you
![]() Delete
all prokaryotic sequences from testseq5.aln using clustalx
Delete
all prokaryotic sequences from testseq5.aln using clustalx
![]() Save
as phylip file (testseq5b.phy)
Save
as phylip file (testseq5b.phy)
![]() Use
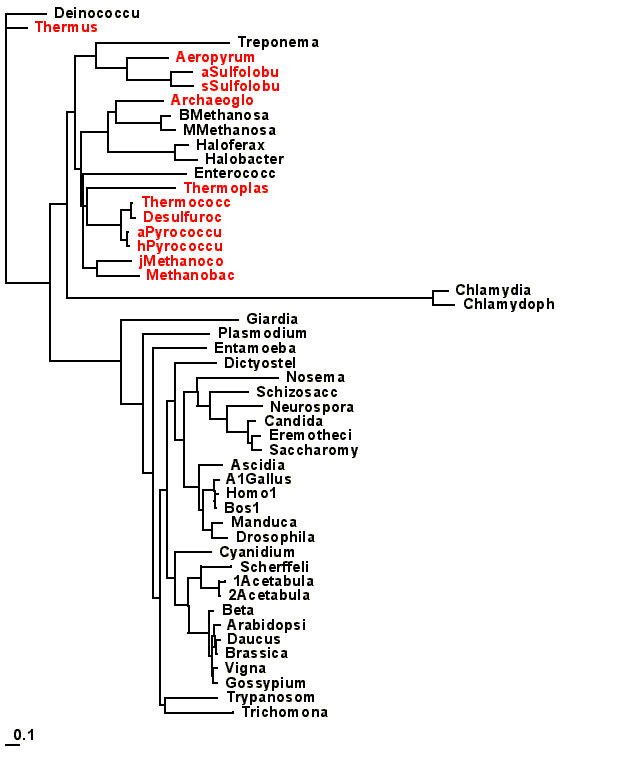
the treeview editor to build the appropriate trees to test. A good starting tree is this
one [it is a neighbor joining tree calculated from distances that were
estimated using among site rate variation (gamma distribution with alpha=.6)
and the JTT substitution model. I
then collapsed all branches that in a maximum likelihood reconstruction were
not at least 2.5 standard deviations larger than zero.] You can load several trees into TREE-PUZZLE simultaneously. If you do so, TREE-PUZZLE also performs a Kishino-Hasegawa
test to determine which of the trees is significantly better than the other
ones. An example for a file with multiple
user trees is here. You can use treeview to
edit your tree to generate the different starting trees. You can copy directly from treeview and paste
into a text editor. The only problem is that treeview adds ‘ ‘
to sequences that start with a number e.g. ‘1Acetabularia’. You need to remove the ‘s before you can read
the trees into TREE-PUZZLE!
Use
the treeview editor to build the appropriate trees to test. A good starting tree is this
one [it is a neighbor joining tree calculated from distances that were
estimated using among site rate variation (gamma distribution with alpha=.6)
and the JTT substitution model. I
then collapsed all branches that in a maximum likelihood reconstruction were
not at least 2.5 standard deviations larger than zero.] You can load several trees into TREE-PUZZLE simultaneously. If you do so, TREE-PUZZLE also performs a Kishino-Hasegawa
test to determine which of the trees is significantly better than the other
ones. An example for a file with multiple
user trees is here. You can use treeview to
edit your tree to generate the different starting trees. You can copy directly from treeview and paste
into a text editor. The only problem is that treeview adds ‘ ‘
to sequences that start with a number e.g. ‘1Acetabularia’. You need to remove the ‘s before you can read
the trees into TREE-PUZZLE!
![]() Save
the trees (multiple trees should be separated by ; )you want to test in a
textfile in the same directory as the TREE-PUZZLE program
Save
the trees (multiple trees should be separated by ; )you want to test in a
textfile in the same directory as the TREE-PUZZLE program
![]() Start TREE-PUZZLE
Start TREE-PUZZLE
![]() Load
testseq5b.phy
Load
testseq5b.phy
![]() Select
tree search procedure and toggle to “User defined trees”
Select
tree search procedure and toggle to “User defined trees”
![]() Select
model of heterogeneity (in the interest of time you might leave it at the
default setting)
Select
model of heterogeneity (in the interest of time you might leave it at the
default setting)
![]() Calculate
branch lengths and likelihood values for the different trees.
Calculate
branch lengths and likelihood values for the different trees.
![]() Use
the CHI square distribution (see handout, or Paul's Chi square calculator
here,
or here) to determine, if the increase in likelihood (2(logL1-logL2)) is significant.
Use
the CHI square distribution (see handout, or Paul's Chi square calculator
here,
or here) to determine, if the increase in likelihood (2(logL1-logL2)) is significant.
B)
Does the incorporation of among site rate
variation lead to a significant increase in likelihood?
For this you
![]() Delete
all prokaryotic sequences from testseq5.aln using clustalx
Delete
all prokaryotic sequences from testseq5.aln using clustalx
![]() Save
as phylip file (testseq5b.phy)
Save
as phylip file (testseq5b.phy)
![]() Save this treefile (see above) in your TREE-PUZZLE directory
Save this treefile (see above) in your TREE-PUZZLE directory
![]() Start TREE-PUZZLE
Start TREE-PUZZLE
![]() Load
testseq5b.phy
Load
testseq5b.phy
![]() Select
tree search procedure and toggle to “User defined trees”
Select
tree search procedure and toggle to “User defined trees”
![]() Select
the default model of heterogeneity
Select
the default model of heterogeneity
![]() start the program
start the program
![]() when prompted enter name of treefile
when prompted enter name of treefile
![]() When done, save outfile under a different name!
When done, save outfile under a different name!
![]() run TREE-PUZZLE again, but select a different model for
substitution (Gamma with 8 categories and alpha=.6)
run TREE-PUZZLE again, but select a different model for
substitution (Gamma with 8 categories and alpha=.6)
![]() Use
the CHI square distribution to determine if the increase in likelihood between the two runs (2(logL1-logL2)) is significant.
Use
the CHI square distribution to determine if the increase in likelihood between the two runs (2(logL1-logL2)) is significant.
Does
{kind=link}