Part I.
After performing blastp search against nr with the unknown ORF as query,
the following results was obtained:
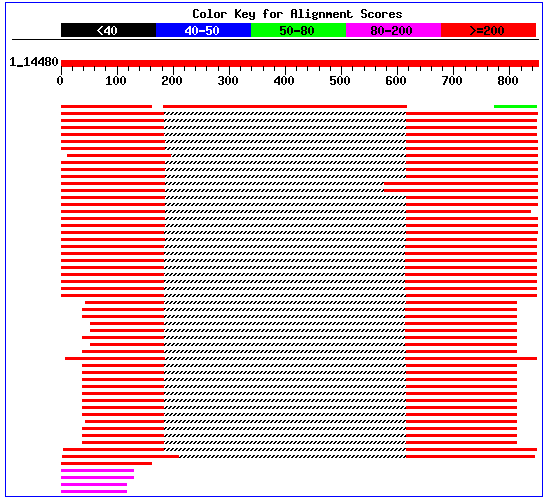
From the depicted graphic overview, it is clear that there is no match that corresponds to the whole length of the query sequence. Rather there is a match to the middle of one protein (the best hit, top row), and the rest of the top hits only have matches to the beginning and the end of the query sequence. (The gray shaded area in the middle corresponds to "no match at all"). The latter matches (the ones that align to the beginning and the end) are from PROBABLE TRANSCRIPTION TERMINATION FACTOR RHO.
The middle portion of the top hit corresponds to part of clpP subunit of Clp protease in Chlamydomonas eugametos:
Query: 184 KCLTSDFHTVLTTRGFIPIADVTLDDKVAVLDNNTGDMSYQNPQKVHKYDYDGPMYDVKT 243
+CLTSD HTVLTTRG+IPIADVTLDDKVAVLDNNTG+MSYQNPQKVHKYDY+GPMY+VKT
Sbjct: 447 ECLTSD-HTVLTTRGWIPIADVTLDDKVAVLDNNTGEMSYQNPQKVHKYDYEGPMYEVKT 505
Query: 244 AGVELFVTPNHRMYVXXXXXXXXQNGYNLVEASSIFGKKVRYKNDAIWNKTDYQFILPDT 303
AGV+LFVTPNHRMYV YNLVEASSIFGKKVRYKNDAIWNKTDYQFILP+T
Sbjct: 506 AGVDLFVTPNHRMYV-NTTNNTTNQNYNLVEASSIFGKKVRYKNDAIWNKTDYQFILPET 564
Query: 304 ATLTGHTNKISSTPAIQPDMNAFL--FGLWIANG---KIADKTAENNQQKQRWKVILTQV 358
ATLTGHTNKISSTPAIQP+MNA+L FGLWIANG KIA+KTAENNQQKQR+KVILTQV
Sbjct: 565 ATLTGHTNKISSTPAIQPEMNAWLTFFGLWIANGHTTKIAEKTAENNQQKQRYKVILTQV 624
Query: 359 KEDVCEIIEQTLNKLGFNFIRSGKDYTIENKQLFSYLNPFENGALNKYLPDFA-DLTT-- 415
KEDVC+IIEQTLNKLGFNFIRSGKDYTIENKQL+SYLNPF+NGALNKYLPD+ +L++
Sbjct: 625 KEDVCDIIEQTLNKLGFNFIRSGKDYTIENKQLWSYLNPFDNGALNKYLPDWVWELSSQQ 684
Query: 416 CKIL-----------TKASNKAYYFSTSERFANDVSRLALSASHAGTTSTGGLDAAPSNL 464
CKIL TK + +YFSTSERFANDVSRLAL HAGTTST L+AAPSNL
Sbjct: 685 CKILLNSLCLGNCLFTKNDDTLHYFSTSERFANDVSRLAL---HAGTTSTIQLEAAPSNL 741
Query: 465 YDTIIGLPTMERVTEYRVIVNQSSFFSYSTDKTTVLNLS--------AQSALSLEQNSQK 516
YDTIIGLP T +RVI+NQSSF+SYSTDK++ LNLS AQSAL+LEQNSQK
Sbjct: 742 YDTIIGLPVEVNTTLWRVIINQSSFYSYSTDKSSALNLSNNVACYVNAQSALTLEQNSQK 801
Query: 517 INKNTLVLTKNNVKSQT-HSQRAERWDTALLTQKELDNSLNHDILINKNPGTSQLECVVN 575
INKNTLVLTKNNVKSQT HSQRAER DTALLTQKELDNSLNH+ILINKNPGTSQLECVVN
Sbjct: 802 INKNTLVLTKNNVKSQTMHSQRAERVDTALLTQKELDNSLNHEILINKNPGTSQLECVVN 861
Query: 576 PEVNNTSTNDRFVYWKGPVYSLTGPNNVFYVQR-GKAVWTHNT 617
PEVNNTSTNDRFVY+KGPVY LTGPNNVFYVQR GKAVWT N+
Sbjct: 862 PEVNNTSTNDRFVYYKGPVYCLTGPNNVFYVQRNGKAVWTGNS 904
NOTE that the match above does not go along the whole sequence,
but corresponds only to the middle part of both query and subject sequences.
If one looks at the annotation of GenBank record of the subject sequence
(gi|1168975), it says that "Region: 448..903 /region_name="Mature chain"
/note="CEU CLPP INTEIN."". This region corresponds exactly to the subject
sequence. The middle portion of the query sequence does NOT match to the
clp protease, but to an intein present in this particular clp protease.
As an alternative, a PSI-BLAST search could be performed to detect the
function of the middle part (different inteins will be returned as the
results of the search).
What might be the function(s) of this protein?
The protein in question appears to be a mosaic protein; Part of it is
homologous to transcription termination factor Rho; but it also has an
intervening sequence, an intein that is homologous to the intein present
in Clp protease in Chlamydomonas eugametos.
Is there a common functional or structural feature among the homologous
proteins?
No. The different homologous proteins are homologous to different parts
of the query sequence, and hence do not have a common functional or structural
feature.
What does this tell you about the protein encoded by this ORF?
It is a protein that has been invaded by an intein.
Which catalytic activity(/ies) would you expect for the encoded protein?
One might perform the CD search:
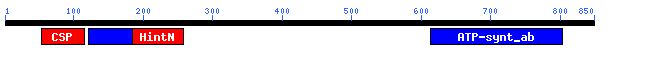
The returned domains are HintN (domain found in inteins), other domains
are Nucleotide binding domains and Cold Shock Protein (RNA-binding) domain.
Intein part: it is a large intein and it has endonuclease activity
(see http://www.neb.com/inteins/Ceu_ClpP.html)
Extein part: nucleotide binding via Rossman fold (homologous to
ATPase catalytic and non-catalytic sbunits) and RNA binding.
To find out if the extein is an ortholog to the Rho termination
factor (and thus probably functions as a Rho termination factor), or if
the extein is a paralog to the Rho termination factor (and probably
has a different cellular function), one has to reconstruct the phylogeny
of the Rho termination factor, its paralogs and the extein portion of
the query sequence.
To do so, one can perform a BLAST search with an extein part of the query
sequence, align the sequences in ClustalX, and then calculate a phylogeny.
As we used a datafile in class that contained the different homologs to
the ATPase catalytic subunits and rho termination factors (testseq2),
the easiest would have been to add the unknown ORF (with the intein removed
if you want to make sure that the alignment works without a problem),
and a couple of rho termination factors to the testseq2-file.
The resulting phylogenetic reconstruction (clustalx) shows different
clusters for the proteins with different function (A/V-ATPase catalytic
subunits in red, F-ATPase catalytic subunits in blue, ..., rho termination
factors in turquoise):
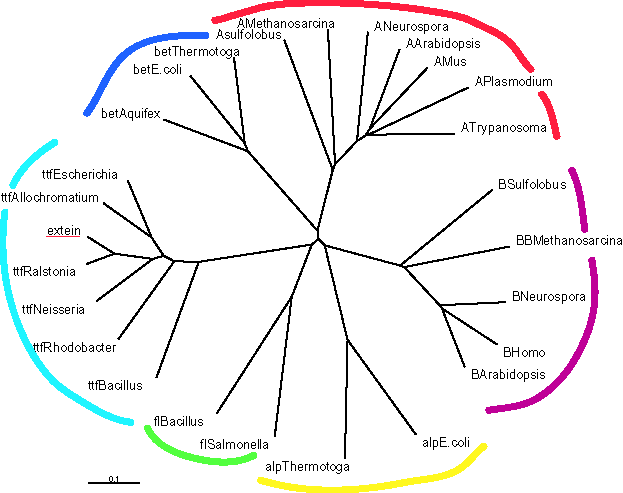
The extein from the unknown ORF groups within the group of Rho termination factors, and hence most probably is an ortholog to the Rho termination factor.
Part II.
Do all of the bacterial sequences (Chlamydia, Chlamydiophila, Enterococcus
(f and hirae), Thermus, Deinococcus, pnStreptococcus (pneumoniae), pyStreptococcus
(pyogenes), Clostridium, Treponema (1&2) and Borrelia) group together?
No
Depicted below is a Bootstrap consensus tree of a Neighbor-Joining Analysis
performed in ClustalX.
Positions with gaps were excluded from analysis, and correction for multiple
substitution was used. The Bacteria are underlined in red. 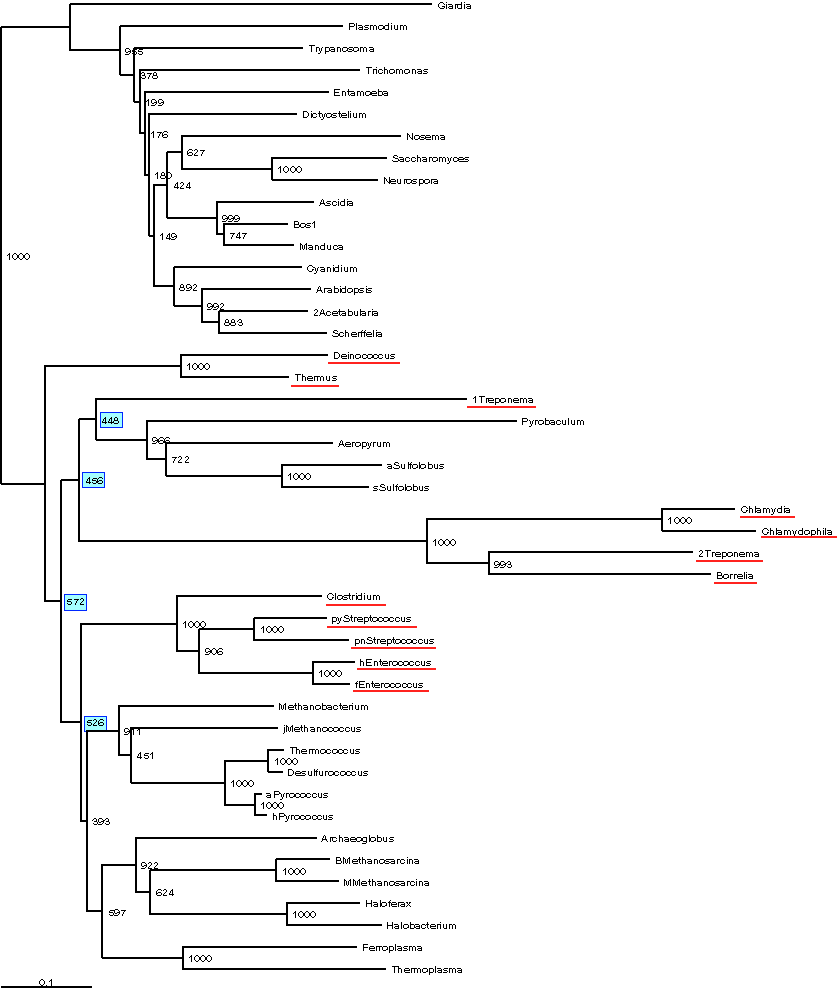
Another phylogenetic reconstruction was performed using TREE-PUZZLE. Most
of eukaryotic sequences were removed from the analysis, since they formed
a strongly supported single group. Below is a puzzling tree with ASRV
taken into account (shape parameter a=0.6).
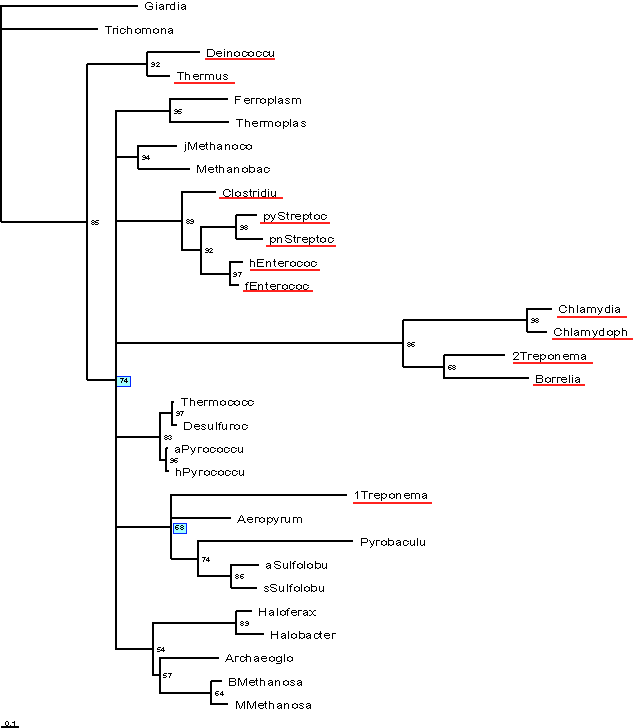
Again, the bacteria do not form a single group.
Another possible analysis would be to calculate a bootstrap analysis using protein parsimony as implemented in PHYLIP. The section below is copied from the outfile. Again, most of the eukaryotes were deleted, as were the bacterial sequences that constitute long branches in the above analyses. All gaps in the input file were replaced by "?". The following programs were used in sequence: SEQBOOT, PROTPARS, CONSENSE. Below is a section from the outfile from CONSENSE:
Consensus tree program,
version 3.6a2.1
Species in order:
1. Giardia
2. Trichomona
3. Archaeoglo
4. MMethanosa
5. BMethanosa
6. Halobacter
7. Haloferax
8. Thermoplas
9. Ferroplasm
10. Methanobac
11. Aeropyrum
12. sSulfolobu
13. aSulfolobu
14. jMethanoco
15. hPyrococcu
16. aPyrococcu
17. Desulfuroc
18. Thermococc
19. Clostridiu
20. fEnterococ
21. hEnterococ
22. pnStreptoc
23. pyStreptoc
24. Thermus
25. Deinococcu
Sets included in the consensus tree
Set (species in order) How many times out of 100.00
.......... .......... .**.. 100.00
.......... ....****.. ..... 100.00
...**..... .......... ..... 100.00
.......**. .......... ..... 100.00
.......... .......... ...** 100.00
.......... ***....... ..... 100.00
.......... .........* *.... 100.00
.......... ......**.. ..... 100.00
.......... .**....... ..... 100.00
..******** ********** ***** 100.00
.......... ........** ***.. 100.00
.....**... .......... ..... 100.00
.......... .........* ***.. 99.00
.......... ....**.... ..... 99.00
..******** ********** ***.. 91.33
.........* ...*...... ..... 74.20
..*****... .......... ..... 71.10
.......... ***.****.. ..... 64.50
.........* ********.. ..... 61.90
.......**. ........** ***.. 60.20
.......*** ********** ***.. 45.50
..***..... .......... ..... 43.80
Sets NOT included in consensus tree:
Set (species in order) How many times out of 100.00
..*******. ........** ***.. 31.00
..*..**... .......... ..... 27.50
.........* ********** ***.. 22.50
..***..*** ********** ***.. 19.00
...****... .......... ..... 17.20
.......... ****...... ..... 13.00
//stuf deleted//
.......... ........** ***** 3.17
Extended majority rule
consensus tree
CONSENSUS TREE:
the numbers on the branches indicate the number
of times the partition of the species into the two sets
which are separated by that branch occurred
among the trees, out of 100.00 trees
+------Desulfuroc
+100.0-|
| +------Thermococc
+100.0-|
| | +------hPyrococcu
| +-99.0-|
+-64.5-| +------aPyrococcu
| |
| | +-------------Aeropyrum
| +100.0-|
+-61.9-| | +------sSulfolobu
| | +100.0-|
| | +------aSulfolobu
| |
| | +------Methanobac
| +---------------74.2-|
+-45.5-| +------jMethanoco
| |
| | +------Thermoplas
| | +--------------100.0-|
| | | +------Ferroplasm
| | |
| | | +------pnStreptoc
| +-60.2-| +-100.0-|
| | | +------pyStreptoc
+-91.3-| | +-99.0-|
| | | | | +------hEnterococ
| | +100.0-| +100.0-|
| | | +------fEnterococ
| | |
| | +--------------------Clostridiu
| |
| | +------Halobacter
| | +--------100.0-|
+100.0-| | | +------Haloferax
| | +---------------71.1-|
| | | +------MMethanosa
| | | +100.0-|
| | +-43.8-| +------BMethanosa
| | |
+------| | +-------------Archaeoglo
| | |
| | | +------Deinococcu
| | +-----------------------------------100.0-|
| | +------Thermus
| |
| +-------------------------------------------------------Trichomona
|
+--------------------------------------------------------------Giardia
remember: this is an unrooted tree!
Can you confidently exclude this possibility?
Based on the bootstrapped NJ tree - No, since the bootstrap support values
for different bacterial groups are very low (the bootstrap support values
are highlighted in blue)
Based on TREE-PUZZLE tree- Still no, although the support values for some
groups became better.
Based on the parsimony analysis - Maybe "yes", but only with
moderate confidence: The Deinococcus -Thermus group is separated
from the other bacterial by a branch with 91.3% bootstrap support. However,
all of the bacteria group together in 3.17% of the bootstrapped samples.
Does the answer change, if one removes the sequences associated with
very long branches?
No
The following long branches were removed: 1Treponema, 2Treponema, Borrelia,
Pyrobaculum, Chlamydia, Chlamydophila. The results of TREE-PUZZLE run
with pruned dataset are below:
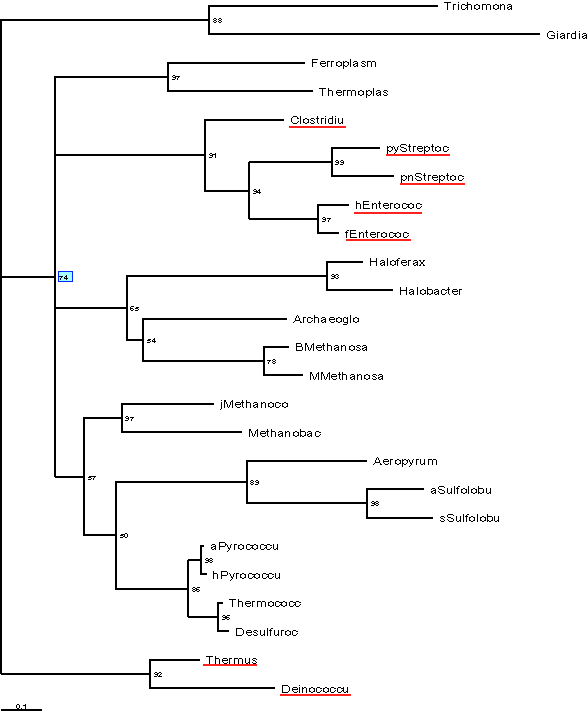
Still cannot confidently exclude the possibility of the bacterial sequences
to form a single group.
Does your answer change, if you consider among site rate variation?
No. See Tree-Puzzle analyses above.
Using ML-mapping the results are a little more decisive. The four clusters
were chosen in way so that according to the puzzle and clustalx trees
the bacteria would be split into two groups that group with the other
two clusters.
Try #1: First we try the Deinococcus/Thermus group as distinct from pyStreptoc, pnStreptoc, hEnterococ, fEnterococ, Clostridium:
LIKELIHOOD MAPPING ANALYSIS
Number of quartets: 260 (all possible)
Quartet trees are based on approximate maximum likelihood values
using the selected model of substitution and rate heterogeneity.
Sequences are grouped in 4 clusters.
Cluster a: 2 sequences
Deinococcu
Thermus
Cluster b: 13 sequences
Thermococc
Desulfuroc
aPyrococcu
hPyrococcu
jMethanoco
Methanobac
Ferroplasm
Thermoplas
BMethanosa
MMethanosa
Haloferax
Halobacter
Archaeoglo
Cluster c: 5 sequences
pyStreptoc
pnStreptoc
hEnterococ
fEnterococ
Clostridiu
Cluster d: 2 sequences
Trichomona
Giardia
Quartets of sequences used in the likelihood mapping analysis are generated
by drawing one sequence from each of the clusters a, b, c, and d.
LIKELIHOOD MAPPING STATISTICS
Occupancies of the three areas 1, 2, 3:
(a,b)-(c,d)
/\
/ \
/ \
/ 1 \
/ \ / \
/ \ / \
/ \/ \
/ 3 : 2 \
/ : \
/__________________\
(a,d)-(b,c) (a,c)-(b,d)
Number of quartets in region 1: 42 (= 16.2%)
Number of quartets in region 2: 8 (= 3.1%)
Number of quartets in region 3: 210 (= 80.8%)
Occupancies of the seven areas 1, 2, 3, 4, 5, 6, 7:
(a,b)-(c,d)
/\
/ \
/ 1 \
/ \ / \
/ /\ \
/ 6 / \ 4 \
/ / 7 \ \
/ \ /______\ / \
/ 3 : 5 : 2 \
/__________________\
(a,d)-(b,c) (a,c)-(b,d)
Number of quartets in region 1: 39 (= 15.0%) left: 24 right: 15
Number of quartets in region 2: 5 (= 1.9%) bottom: 4 top: 1
Number of quartets in region 3: 199 (= 76.5%) bottom: 76 top: 123
Number of quartets in region 4: 4 (= 1.5%) bottom: 2 top: 2
Number of quartets in region 5: 3 (= 1.2%) left: 3 right: 0
Number of quartets in region 6: 6 (= 2.3%) bottom: 5 top: 1
Number of quartets in region 7: 4 (= 1.5%)
Only 3.1% of the quartets support a single bacterial group, and only 1.2% do so strongly. In contrast the split of the bacterial sequences into two groups is strongly supported by 199 quartets (=76.5%).
Try #2: The clusters are chosen to ask the question: does 1Treponema
group with the Crenarchaeotic sequences?
LIKELIHOOD MAPPING ANALYSIS
Number of quartets: 260 (all possible)
Quartet trees are based on approximate maximum likelihood values
using the selected model of substitution and rate heterogeneity.
Sequences are grouped in 4 clusters.
Cluster a: 1 sequences
1Treponema
Cluster b: 13 sequences
Thermococc
Desulfuroc
aPyrococcu
hPyrococcu
jMethanoco
Methanobac
Ferroplasm
Thermoplas
BMethanosa
MMethanosa
Haloferax
Halobacter
Archaeoglo
Cluster c: 5 sequences
pyStreptoc
pnStreptoc
hEnterococ
fEnterococ
Clostridiu
Cluster d: 4 sequences
aSulfolobu
sSulfolobu
Aeropyrum
Pyrobaculu
Quartets of sequences used in the likelihood mapping analysis are generated
by drawing one sequence from each of the clusters a, b, c, and d.
LIKELIHOOD MAPPING STATISTICS
Occupancies of the three areas 1, 2, 3:
(a,b)-(c,d)
/\
/ \
/ \
/ 1 \
/ \ / \
/ \ / \
/ \/ \
/ 3 : 2 \
/ : \
/__________________\
(a,d)-(b,c) (a,c)-(b,d)
Number of quartets in region 1: 0 (= 0.0%)
Number of quartets in region 2: 28 (= 10.8%)
Number of quartets in region 3: 232 (= 89.2%)
Occupancies of the seven areas 1, 2, 3, 4, 5, 6, 7:
(a,b)-(c,d)
/\
/ \
/ 1 \
/ \ / \
/ /\ \
/ 6 / \ 4 \
/ / 7 \ \
/ \ /______\ / \
/ 3 : 5 : 2 \
/__________________\
(a,d)-(b,c) (a,c)-(b,d)
Number of quartets in region 1: 0 (= 0.0%) left: 0 right: 0
Number of quartets in region 2: 20 (= 7.7%) bottom: 20 top: 0
Number of quartets in region 3: 218 (= 83.8%) bottom: 171 top: 47
Number of quartets in region 4: 0 (= 0.0%) bottom: 0 top: 0
Number of quartets in region 5: 22 (= 8.5%) left: 14 right: 8
Number of quartets in region 6: 0 (= 0.0%) bottom: 0 top: 0
Number of quartets in region 7: 0 (= 0.0%)
7.7% of the quartets group 1 Treponema with the other bacteria. Removing long sequneces form cluster b might improve the result further.
Based on results of ML mapping analyses, especially try #1, the tentative
conclusion is that the bacterial A-type ATPase catalytic sequences do
not form a single group.
What is the implication of your analysis?
Within the Eukaryotes and within the Archaea the phylogeny of the V/A-ATPases agrees very nicely with the evolution of other conserved molecular markers (rRNA, elongation factors). The F-ATPase bacterial F-ATPases also were found to agree with rRNA to a surprising extend. In contrast the A/V-ATPase subunits found in some bacteria have a phylogeny that does not agree well with other markers, and one indication is that these bacterial subunits do not form a strongly supported group compared to the Archaeal and Eucaryal homologs.
The presence of archaeal type ATPases in bacteria can be explained two
ways:
A) these archaeal ATPases were transferred into the bacterial domain by
horizontal gene transfer. The placement of these ATPases relative to the
archaeal and eukaryotic homologs would then indicate the position of the
donating organism.
B) the last common ancestor of archaea and Bacteria had both the F- and
the A/V- type ATPase and the current distribution of ATPases is explained
through gene loss. In this case, the phylogeny of the A-ATPases should
reflect organismal evolution.
The performed analysis indicate that horizontal gene transfer impacted the distribution of A-ATPases among bacteria, but more analysis need to be done to test different alternatives. For example, at present it remains unclear if the A-ATPases in the Deinococcaceae and the Borrelia and Chlamydia ATPases have resulted from a single transfer event. Methods one might use include ml ratio tests and the calculation of posterior probabilities.