These are exercises and for the most part not
questions to be answered. The ones that
you should answer are in pink:
1. Getting to know SPDBV
Save
1HEW.pdb onto your computer - Open in chime (from Netscape) and save pdb
file from the chime files menu or by long-clicking
here –
if you choose the later option, you need to open the file in word and save it
under a different name in text format)
Start SPdbV
load
1HEW.pdb (hit return)
click
the right mouse button (= sign on Mac, in most other instances the alt-key together
with the mouse-click has the same effect as the right button on the PC mouse)
to center the molecule
click
on the three cursor control buttoms and rotate/move/enlarge lysozyme picture
click
on the page icon and go through the pdb file
open
the control window (Window -menu).
open
the align window (Window -menu)
select
all (Select menu)
Window
Ramachandran plot
(In case you forgot what this is about, go here.)
in
the control or the align window select different residues
select
all (Select menu)
Explore
different coloring schemes (CPK, secondary structure, accessibility) and
display options (show CA trace only, show oxygen, …)
REMARK: If you do serious
work save your work periodically, sometimes it is impossible to recover from an
inadvertent mouse click)
Select
the NAG inhibitor (go to the at the bottom of the control panel, click with
left mouse button).
Color CPK
Invert
selection (return) (Select menu)
Color secondary structure
Invert
selection (return)
Tools calculate H-Bonds
right
click inside the side chain column in the DISPLAY window to turn off side chain
display
select
Neighbors of selected aa (5 to 10 Angstrom is appropriate) (Select menu)
hit
return
click
on side-heading in control panel (clicking in the header acts only on
selected residues), add labels, change color ….
select
group properties non-polar aa (Select menu)
click
on Header COL in control panel select blue color to color hydrophobic
residues blue
Are there “blue” residues interacting with the N-Acetyl
glucosamines? How come? (Aren’t
carbohydrates supposed to be hydrophilic?)
The
resulting display after some manipulations might look like this (after you save
it as a pov-ray image):
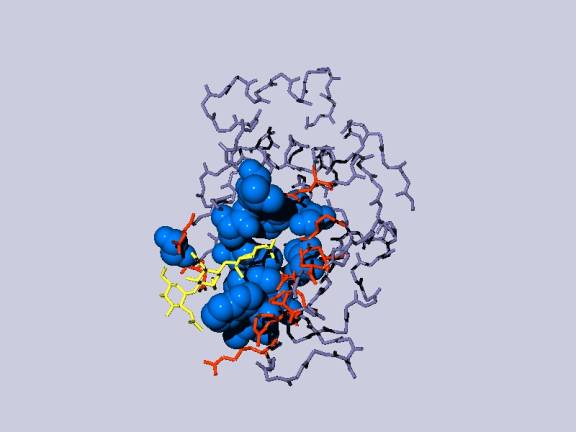
yellow:
the NAG inhibitor;
blue: residues in the binding pocket that are non-polar, depicted as space
filling balls;
red: other amino acids in the binding
pocket;
gray: the rest of the Lysozyme molecule, but only the backbone.
Play
around, if in doubt use the ? buttom.
The
worst that can happen is that you'll have to restart your computer.
Open
the alignment window and display the complete lysozyme molecule. Observe the color change in the structure
that happens when you move the mouse over the sequence in the alignment
window.
Other
things to try:
3D rendition (in the display menu),
slab view (shift and mouse move the slab),
explore the make up of the pdp file (text icon below the cursor
control),
have a look at the opening control window (upper left icon below
the cursor control).
If
you right click on a residue in either the alignment window or the control
window, the display centers on this residue.
Control
and mouse click adds residues to the list of selected residues (works in either
window)
2. Exercise on
multi subunit proteins:
Aligning F-ATPase alpha and beta subunits
Start SPdbV
Open 1bmf.pdb (save from here - remember
the text file re-saving from above -, or open with chime – netscape - and save
from file menu)
(F-ATPases are typically found in bacteria and mitochondria and plastids. They either function as proton pumping ATPases, or they use the
proton gradient to synthesize ATP. They
can be dissociated into two parts; the F1 portion binds and hydrolyses
ATP. The conformation change associated
with this catalytic cycle of ATP binding, hydrolyses and ADP+Pi dissociation is
communicated to the membrane imbedded FO-portion via the gamma
subunit (part of F1). Click here for
animated gifs displaying the catalytic cycle, and here for a
movie of the actual rotation as seen under the microscope)
Color Chain
Change color chainD to
grey/blue (to do this right click in control panel on D in first column to
select chain D, right click on COL, select color)
Scroll down the control
panel and select all ATP analogs (press ctrl key and right click to select)
right click on COL in
heading and select red color
Read the pdb file to get
info on which chain is which
select chain F (including
nuc) and save selected residues as betaTP.
select chain A (including
nuc) and save selected residues as alphaE.
After playing with the
F1-ATPase, close this file and open betaTP and alphaE.
Display layer info (windows
menu)
To avoid confusion select
and display the nucleotides only
There are
different ways to align 3-D structures.
One way is to select 3 corresponding points in each of the two
structures. To do so you can use the substrate molecule.
Using the mov check off in
the Layer Info, reorient the two ANPs so that they are in a similar orientation
(but not overlapping).
Click on the align bottom
with the 3 green and 3 red dots. Notice
the red instructions that appear in the header next to the pdb-page icon. Follow these instruction using three
corresponding atoms.
SHIFT DISPLAY Show CA trace
only (Shift makes the commands act on both layers)
Using the mov checks in the
Layer info, move the two chains next to each other.
What do you think about the
result?
Another way to align
structures is to use the magic fit in
the tools command. Do this and run
improve fit (notice the red info in the header)
Click on alpha in Layer
info to make the alpha subunit the active layer
Color CPK
Make the beta subunit the
active layer
COLOR rms . The further the
atoms in the beta subunit are away from the alpha subunit, the longer
wavelengths it is the colored.
DISPLAY Show alignment
window - gives you the aligned sequences.
Use the mouse
over function to find the parts of the structure which are used in the
alignment, and the part that is not used.
What could one do to get an alignment of the carboxy portion parts that
is not aligned after magic fit?
How does this
alignment compare to the ones calculated using Clustalw? (You can save your alignment as a
text file from the File menu; testseq2 in Assignments4 contained both beta and
alpha F-ATPase subunits.)
If you have time, repeat
the exercise for the four histones that are part of the nucleosome (see lecture
notes on class 5).