Why phylogenetic reconstruction of molecular evolution?
A. Systematic
classification of organisms
e.g.: Who were the first angiosperms? (i.e. where are the first
angiosperms located relative
to present day angiosperms?) Where
in the tree of life is the last common ancestor located?
B. Evolution
of molecules
e.g.: domain shuffling,
reassignment of function, gene duplications, horizontal gene transfer
By which organisms and from what precursors was
eukaryotic DNA packaging invented?
What are the roots of the eukaryotic cytoskeleton?
C. Mechanisms
of evolution
e.g.: What is the role of
horizontal gene transfer in microbial evolution?
Are speciation events correlated
with or caused by major genome rearrangements?
Did duplications of whole genomes
provide the material that allowed more complex developmental pathways to
evolve?
How:
1)
Obtain sequences
Sequencing
Databank Searches -> ncbi a)
entrez, b) BLAST, c) blast of pre-release data
Friends
2)
Determine homology (practically you only can determine the non-randomness of a
match, but at present most believe that this is a sufficient demonstration of
homology)
Recall the following definition:
Homology:
two sequences are homologous, if there existed an ancestral molecule in
the past that is ancestral to both of the sequences
Types of homology:
Orthology:
bifurcation in molecular tree reflects
speciation
Paralogy: bifurcation in molecular tree reflects gene
duplication
Xenology: gene was obtained by organism through
horizontal transfer
Synology: genes ended up in one organism through fusion
of lineages.
3)
Align sequences
(most algorithms used for phylogenetic reconstruction require a global
(as opposed to a pairwise) alignment exception:
statalign from Thorne JL, and Kishino H, 1992, Freeing phylogenies from
artifacts of
alignment. Mol Bio Evol 9:1148-1162)
a.
algorithms
doing a global alignment: clustalw 1.7, or pile_up (GCG)
b.
local
alignments (MACAW)
4)
Reconstruct evolutionary history
a. Distance Analyses
- calculate pairwise
distances
(different distance measures, correction for multiple hits, correction for codon bias) - make distance matrix
(table of pairwise corrected distances)
- calculate tree from
distance matrix
b. Parsimony Analyses
find that tree that explains sequence data with minimum number of
substitutions
(tree includes hypothesis of sequence at each of the nodes (acctrans,
deltrans)
c. Maximum
Likelihood Analyses
given a model for sequence evolution, find the tree that has the highest
probability under this model.
This approach can also be used to successively refine the model.
Else:
spectral analyses (i.e., look at patterns of substitutions): evolutionary
parsimony, Hadamard conjugation
Another way to categorize methods of phylogenetic
reconstruction is to ask if they are using
(i) an optimality criterion (e.g.: smallest error
between distance matrix and distances in tree, least number of steps), or
(ii) algorithmic approaches (UPGMA or neighbor joining)
Orthologues: bifurcation in
molecular tree reflects speciation
– these are the molecules people interested in the taxonomic classification of
organisms want to study
Paralogues: bifurcation in
molecular tree reflects gene duplication
The study of paralogues and
their distribution in genomes provides clues on the way genomes evolved. Gen and genome duplication have emerged as
the most important pathway to molecular innovation, including the evolution of
developmental pathways.
Xenologues: gene was obtained by
organism through horizontal transfer.
The classic example for Xenologs are antibiotic resistance genes, but
the history of many other molecules also fits into this category:
inteins, selfsplicing introns, transposable elements, ion pumps, other
transporters,
Synologues: genes ended up in one
organism through fusion of lineages.
The paradigm are genes that were transferred into the eukaryotic cell
together with the endosymbionts that evolved into mitochondria and plastids
How
can genes get duplicated: Whole genome duplication, partial genome duplication,
single genes get duplicated (tandem repeats)
Whole
genome duplication: frequent event in plants, also speculated to have
occurred at least twice in the early evolution of vertebrates. 15% of the yeast
genome is present in duplicated form, the currently accepted idea is
that there was an ancient duplication followed by rearrangement and gene loss. The idea of genome duplications in early
vertebrate evolution has become very popular, but phylogeny of regulatory
proteins does not support this idea (see here
and here
for pro and here
for contra).
Parts
of chromosomes get duplicated: traces of this seen in
Arabidopsis and Caenorhabditis
Single
genes get duplicated -> gene families originally tandemly replicated
(see the Caeonrhapditis paper above)
How
many different genes are necessary in an organism?
Surprisingly
few necessary, but usually many more present:
Minimum: prokaryotes 500 - 1000, eukaryotes 5000-10000
To
study genomes, one of the best sites is, as usual, the NCBI.
The genome data bank allows access to plasmids, viral, organellar, pro- and
eukaryotic genomes. It includes both
completed genomes and genomes in progress.
Most of the maps and tables are clickable. Try it out, you cannot break things.
For
example, the Borrelia
genome, if you click on the “complete genome”, you get a graphical
representation, further clicks move you down throw several levels to the
nucleotide and encoded amino acid sequence.
If you click on an ORF, you retrieve the sequence followed by an output
of a blast search of this sequence against the nr database. The graphic representation shows you which
part of the ORF generated the match, if you click on the number that represents
the score, you open a new window with the alignment (again with nice graphics
included). If you click on the number
an window with the matching sequence in gb-format opens up. If the ORF is part of a cluster of
putatively orthologous genes, you can get information on the cluster by
clicking on the COGnumber: For example, follow this
link of the CLP protease from Borrelia.
The COG entry contains a lot of intriguing things and links, you can
download all COG belonging to this cluster, you get a “rough” tree indicating
the relationships of members of this COG, and you can download all COG that
have an identical distribution in the reference genomes as the one on whose
page you landed. (Is it a coincidence
that most of the COGs with the CLP protease distribution are chaperones? And
that these do not conform to the canonical organismal phylogeny?)
Going
back to the Borrelia genome, you can easily go to tables listing all ORF,or to
taxtable, which provides an interesting nearest neighbor coloring of the
genome. It is noteworthy that many of
the pink dots are endonucleases (xenologues).
Also, there are many transporters among the odd colored genes.
Another
cool site is Robert L.
Charlebois genome and
bioinformatics site. While it is
not always clear what the beautiful pictures mean, some of the available tools
are obviously great.
Example1:
Using the organism
phylogenies tool, one can quickly generate gene contents trees; which (you
may or may not be surprised to find out) usually are quite similar to the classical
three domain taxonomy. The example
below was calculated with a cutoff at a blast score of E=10-8 and a
distance based on gene content. Note
that those bacteria usually considered deep branching (Thermotoga and Aquifex)
are also deep branching in this tree, and that the deepest branch in the
archaea leads to the crenarcheote.
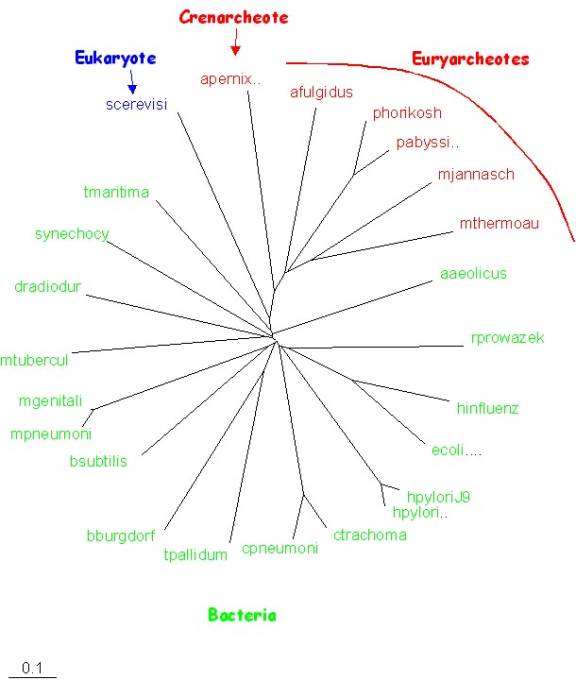
(distances
calculated using Robert L.
Charlebois’ genome and
bioinformatics site, tree calculated with FITCH from the Phylip package
using the default options and global rearrangement and jumbled input)
The
outcome is nothing short of amazing given the strange distance measure used (i.e., the number
of ORFs from the first genome that match something in the second genome better
than the chosen BLASTP cutoff e-value is computed, then divided by the number
of ORFs in the first genome. The distance is then simply 1.0 minus this
proportion (from here); any ideas for a more
appropriate scaling or rescaling? )
Does
this mean 16SrRNA is correct, and all the recent discussions of HGT are
overblown? Or what? What is microbial taxonomy based on?
Using
the genetic mosaicism
program, one can easily search for genes that were imported from outside the
lineage of the organism. For example,
choosing the Deinococcus genome, and entering Archaea and e=-8, one finds genes
whose best blast hit outside the “own lineage” is from the Archaea. Among the many hits are the V/A-ATPase
subunits. How can this finding be
reconciled with the last example???
Genomic dot plots allows to
compare two genomes (or rather the ORF in encoded in these genomes). For example BLASTP-based
dot plot of Pyrococcus abyssi vs Pyrococcus horikoshii depicted below clearly
reveals inversions, and a duplication (two parallel diagonals), the latter can
also be detected by comparing a genome to itself.

The picture below is a comparison of the Yeast proteom with itself (the
diagonal is removed). It clearly shows
many small regions of duplications.

Gene clusters finds
neighboring genes that have the same order in two genomes. Kind of the same as above, but you get a
listing of the clusters and their putative identification. (Example Deinococcus and Methanococcus)